

 PDF
PDF ePub
ePub Citation
Citation Print
Print


INTRODUCTION
The gastrointestinal tract is chronically exposed to various antigens found in bacteria and food. In the normal state in the absence of intestinal inflammation, gut homeostasis is maintained by suppressing excessive immune responses to foreign antigens. IBD is an idiopathic disorder caused by chronic and excessive inflammation of the gastrointestinal tract, leading to rectal bleeding and weight loss.1
2 IBD, a dysregulated immune inflammatory state of the gastrointestinal tract, is classified into 2 archetypal phenotypes, UC and CD. These 2 subtypes of IBD are characterized by chronic inflammation in the gastrointestinal tract and repeated cycles of relapse and remission. Although UC and CD show differences in their clinical presentation, the same risk factors are implicated in the pathogenesis of both subtypes. Phenotypes common to both subtypes include chronic inflammation and a dysregulated immune inflammatory response; therefore, much of the research on IBD pathogenesis has focused on the immune system. The pathogenesis of both UC and CD involve genetic factors, changes in the gut microbiome, and immune response cells including cytokines and immune cells.
Even though the pathogenesis of IBD is complicated, several studies have demonstrated that excessive interleukin (IL)-17 production is involved in the progression of IBD.3 Recently, research on IBD pathogenesis has focused on T helper (Th)17 cells, which secrete IL-17. It is well documented that Th17 inhibition can decrease the development of acute colitis by reducing inflammation.4 Additionally, innate lymphoid cells (ILCs) were recently discovered to be novel pathogenic effector lymphocytes in IBD. In this review, this topic will be discussed primarily in the context of human IBD and experimental IBD animal models. Additionally, current therapeutics targeting Th17 and ILCs will be discussed.
Go to : 
IBD-RELATED CYTOKINES AND CHEMOKINES
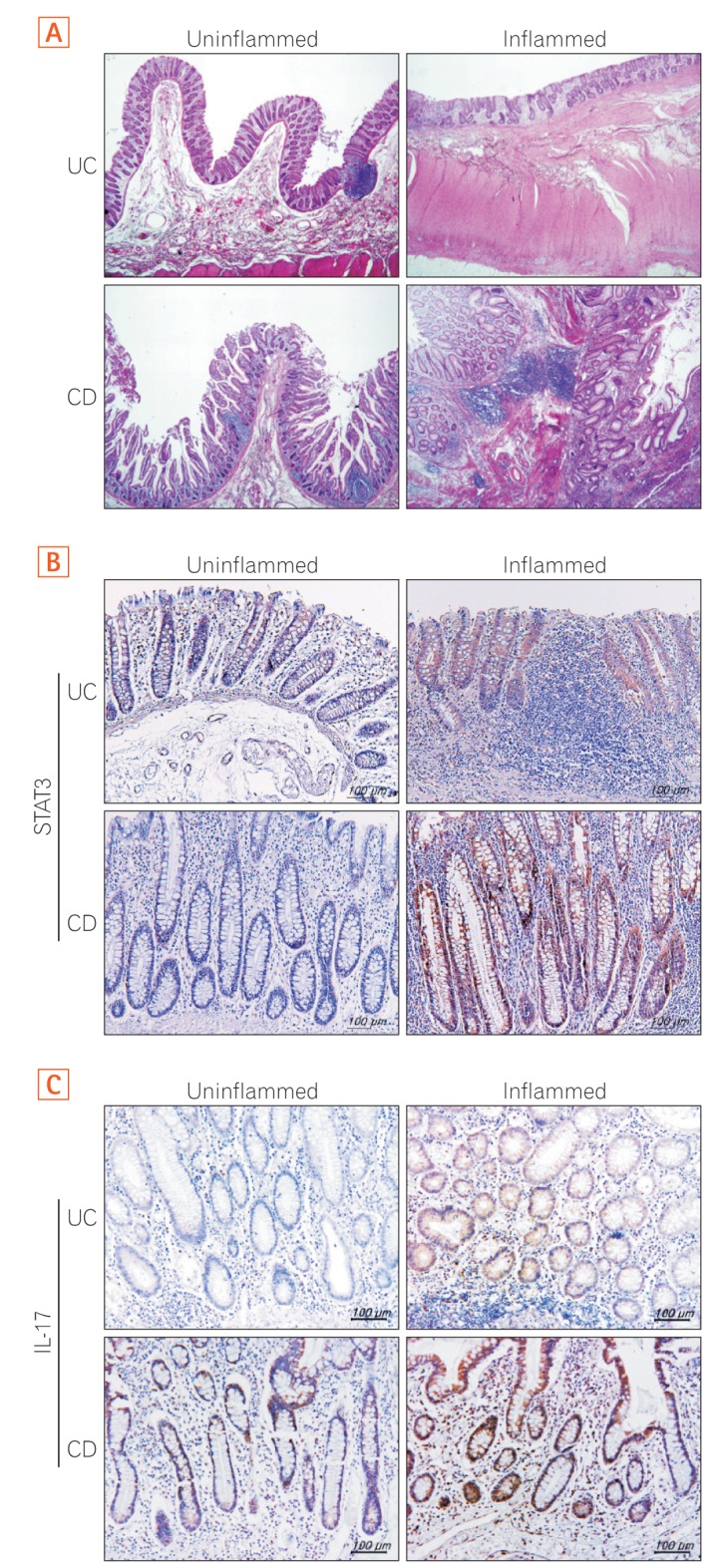
Immune cells secrete products that are actively involved in the initiation and preservation of inflammation, leading to gut tissue damage. In IBD patients, colonic lesions show excessive immune cell infiltration and tissue devastation (Fig. 1A). The expression of STAT3 and IL-17 are also increased in inflamed colon tissue (Fig. 1B and C). Many cytokines and chemokines are associated with IBD development.
1. Cytokines
Several pro-inflammatory cytokines are involved in the progression of IBD. For example, the IL-1 family of cytokines has a key role in IBD pathogenesis.5 In UC, IL-1β promotes inflammation because IL-1 originates from monocytes and macrophages, and active IL-1β is expressed in the colonic mucosa.6 IL-18 is also an IL-1 family member and is increased in the mucosa of CD patients.7 It has been suggested that IL-18 increases the Th1 response.8
9 However, in CD patients with active disease, IL-10 released from mucosal T cells was decreased by IL-18.10 IL-33, another member of the IL-1 family, stimulates mucus secretion to protect the epithelium and upregulates the expression of IL-5 and IL-13 as part of the Th2 response.11 There is evidence that the expression of IL-33 and its receptor ST2 are increased in UC patients.12
13
IL-6 activates signal transducer and activator of transcription 3 (STAT3) and has an important function in the inflammatory response. IL-6 and its soluble IL-6 receptor were increased in UC and CD patients.14
15 IL-6 also has a key role in the pathogenesis of UC and the carcinogenesis of colorectal cancers related to UC.16
Tumor necrosis factor α (TNF-α) has a significant function in IBD pathogenesis because IL-1β, IL-6, and IL-33 expression can all be increased by TNF-α.17
18 The clinical severity of UC and CD were correlated with TNF-α levels in the serum of IBD patients.
IL-10 is a typical immunosuppressive cytokine that may have therapeutic value for treating chronic IBD.19 Although IL-10 is an anti-inflammatory cytokines, there are inconsistencies of IL-10 concentrations in IBD. A study showed that gut IL-10 expression levels were either the same or higher in IBD patients than in normal controls.20 It is well documented that IL-10 gene expression is higher in the mucosal T cells of UC patients than normal controls.21 Furthermore, IL-10 production is enhanced in the serum of CD patients.22 On the other hand, other investigation demonstrated that IL-10 levels in serum of patients with UC and CD are similar to healthy subjects.23 It is also well documented that downregu-lation of IL-10 promotes disease progression in patients with CD.24
25
Transforming growth factor β (TGF-β) has dual activities in the pathogenesis of IBD. It stimulates epithelial compensation and fibrosis and induces tolerance and homeostasis through an impressive immunoregulatory function.26 In the lamina propria, TGF-β1 levels in mononuclear cells were enhanced in UC patients but decreased in CD patients.27 TGF-β improved intestinal inflammation by reducing the expression of IL-33.28
IL-17 is a pro-inflammatory cytokine that activates STAT3, which stimulates a strong chronic immune inflammatory response.29
30 Thus, IL-17 is critical in the pathogenesis of IBD. Indeed, IL-17 mRNA levels were enhanced in the inflamed mucosa of patients with IBD, both UC and CD.31 IL-17 has many isoforms, including IL-17A (also known as IL-17), IL-17B, IL-17C, IL-17D, IL-17E (also known as IL-25), and IL-17F.32 Although IL-17A inhibition can attenuate the inflammatory response, whether IL-17A has a pathogenic role in IBD is controversial. It has been suggested that IL-17A inhibition mediated by phosphorylated STAT3 suppression decreases inflammation and the progression of acute colitis,4 whereas IL-17A can improve experimental colitis.33 Additionally, by reinforcing tight junction formation, IL-17 can also protect human intestinal epithelial cells.34 However, IL-17 is recognized as a significant inflammatory factor in CD pathogenesis. Previous studies found higher levels of IL-17 and CD161+ memory cells expressing IL-17 and interferon γ (IFN-γ) in CD patients.35
36 It has been reported that IL-17 can increase the recruitment of T cells into the lamina propria during the inflammatory response.37
2. Chemokines
IL-8 is mainly a neutrophil chemoattractant that induces the migration of neutrophils from peripheral blood into inflamed tissue. It is well known that IL-8 production is increased in the tissue of UC patients compared with that of normal controls.38 Moreover, other chemokines are elevated in the mucosa of IBD patients. Various reports have shown that the expression of chemokine (C-C motif) ligand (CCL) 2 (also known as monocyte chemoattractant protein [MCP]-1), CCL3 (also known as macrophage inflammatory proteins [MIP]-1α), CCL4 (also known as MIP-1β), CCL7 (also known as MCP-3), CCL20 (also known as MIP-3α), CX-C motif chemokine (CXCL) 5, CXCL8, CXCL10 and regulated on activation, normal T cell expressed and secreted (RANTES) are upregulated in tissues from IBD patients.39
40
41
42
Go to :
Th17 CELLS ARE KEY FACTORS IN THE PATHOGENESIS OF IBD
Presently, Th17 cells are considered a main pathogenic factor in IBD. A study found a massive infiltration of Th17 cells in the inflamed intestinal mucosa of IBD patients.43 Cells that release IL-17 and Th17-related cytokines were also increased in inflamed tissue from IBD patients compared with normal tissue.31
43
1. Th17 Cell Differentiation in the Intestine
T cells can be differentiated into Th17 cells when they interact with various inflammatory mediators in the intestine. Naive T cells can be differentiated into Th1, Th2, Th17, and regulatory T (Treg) cells through a process controlled by effector cytokines produced by antigen-presenting cells. During Th17 polarization, many cytokine receptors (IL-1R, IL-6R, IL-21R, IL-23R, and TGF-βR) of naive T cells have important roles. Many reports have shown that specific cytokines such as IL-1β, IL-6, IL-21, IL-23, and TGF-β induce Th17 polarization.44
45
46 STAT3 was first identified as a Th17 cell specific transcription factor It has been suggested that STAT3 overexpression can promote Th17 differentiation and proliferation. Consistent with this, loss of STAT3 can suppress the differentiation of naive T cells into Th17 cells.47
2. Th17 Cells and the Microbiota
The gastrointestinal tract contains a large microbial community. Thus, gut microbial settlement in the gastrointestinal tract is crucial to the formation of the immune system.48 The microbiota also plays a key role in the pathogenesis of IBD. Indeed, metagenomics research has revealed that various intestinal microbiota genes are involved in host mRNA expression.49 Destruction of the intestinal mucosal barrier is caused by genetic susceptibility in IBD patients.50 Analyses of biopsy samples from UC and CD patients have shown that changes in the composition of the microbiota may be associated with IBD pathogenesis.51
52
53
Communication of T cells with microbes in the gastrointestinal tract is very important for maintaining intestinal immunity. The process of Th17 cell differentiation is determined by factors such as the composition of the endogenous microbiota in the intestine.54
55 Correspondingly, Th17 cell differentiation in the intestine is remarkably decreased in a germ-free system and in the presence of antibiotics.56
57 Additionally, many studies have shown that specific gut microbiota drive Th17 cell differentiation.56
58
3. Th17 Cells Function in Intestinal Inflammation
Th cells are relevant to the pathogenesis of IBD because they are fundamentally plastic to provocation from the surrounding state.59 Th17 cells releasing IL-17 are a strong pro-inflammatory factor in IBD. In a study on IBD patients, IL-17 expression and IL-17A and IL-17F mRNA levels were higher in the mucosa and serum of IBD patients than healthy controls. 31
60 In a subtype of IBD, IL-17 was more abundant in CD patients than UC patients.31 Recently, various cytokines related to Th17 were found to be upregulated in both UC and CD patients compared with normal subjects, but they were higher in the UC patients.61 It has also been documented that UC severity is correlated with IL-17 expression in peripheral blood mononuclear cells.62 Genome-wide association scans showed that various UC and CD susceptibility genes, such as STAT3, were related to Th17 genes; therefore, the Th17 signaling pathway may be involved in the pathogenesis of IBD.63
64
Although it is known that Th17 cells play a role in the pathogenesis of IBD, a dextran sulfate sodium (DSS)-induced colitis model showed that IL-17A and IL-17F have contrasting roles. Studies using this model found that antibody-mediated IL-17A protein suppression and gene knockout caused excessive inflammation and damage to the intestinal epithelium.65
66 On the other hand, in mice with DSS-induced colitis, it was shown that IL-17F could also have a protective role.67 It has also been reported that loss of IL-17F can improve the intestinal inflammatory response.67 Thus, the focus of current IBD therapies is on blocking IL-17A and IL-17F.68
4. Maintenance of Intestinal Th17 and Treg Cell Proliferation
Generally, Th17 and Treg cells modulate the proliferation of each other to maintain balance. It has been reported that the developmental pathways of Th17 and Tregs are related with their differentiation and Th17/Treg balance is important to maintain immune response in intestine.69 This influences the outcome of immune responses in the context of inflammatory conditions.
Go to :
PROTECTIVE FUNCTION OF Treg CELLS IN IBD
Treg cells are associated with the pathogenesis of IBD because the intestinal inflammatory response in IBD is mediated mainly by the T-cell response.70 Treg cells perform a critical role in preserving immune homeostasis and establishing inflammation in response to foreign or non-pathogenic antigens such as commensal bacteria, and failure of Treg cell function can lead to an inflammatory disorder. Indeed, mutations in CD25 and IL-10, which are involved in Treg cell differentiation, lead to aberrant Treg cell function and increased susceptibility to IBD.71 Moreover, loss of IL-10 results in intestinal inflammation, and Treg cells lacking the IL-10 receptor are more susceptible to colitis.72
73
As research using IBD mouse models has shown that Treg cells could suppress intestinal inflammation,74 it is said that Treg cells have an anti-colitis effect. Indeed, ablation of Treg cells or impairment of TGF-β1 signaling in Treg cells increased colitis progression.75
76 Additionally, there is evidence that IL-10 released from Treg cells can decrease colitis progression.77
78
79 In a mouse model, an increase in Treg cell differentiation downregulated the development of experimental ileitis, and co-transfer of conventional T cells and Treg cells decreased intestinal inflammation in a RAG1 knock-out mouse model.80
Go to :
RECIPROCAL BALANCE BETWEEN Th17 AND Treg CELLS IN IBD
As the concentration of T cell differentiation and balance has moved from the Th1/Th2 to that of Th17/Treg, this paradigm has indeed been shown to affect in IBD. Th17 and Treg cells exist primarily in the intestinal mucosa, where they have a significant role in T-cell-mediated immune responses.81
82 It is well known that IL-17-releasing Th17 cells are entirely dependent on STAT3 and are primarily pro-inflammatory.83 On the other hand, Foxp3-expressing Treg cells show anti-inflammatory activity that is mediated through the suppression of the Th17 response. In an experimental colitis model, Treg cells prevented intestinal inflammation and reduced the expression of Th17-related cytokines.84
Go to :
IBD THERAPIES THAT TARGET Th17
As Th17 cells play a key function in intestinal inflammation, it has been proposed that they may be therapeutic targets to regulate the intestinal inflammatory response.
1. Th17 Cell Blockade
Theoretically, pro-inflammatory cytokines, including IL-6, can increase Th17 cell differentiation and proliferation. Therefore, it has been suggested that new IBD therapies may involve neutralizing monoclonal antibodies that target these cytokines or their receptors. It is well documented that monoclonal antibodies against IL-12/23 p40 can improve colitis severity in murine models.85
86 Consistent with this, neutralizing IL-21 antibody treatment downregulated the infiltration of colonic T cells and the expression of pro-inflammatory cytokines such as IL-6 and IL-17A in inflamed intestinal tissue from mice with DSS-induced colitis.87
Potential strategies for IBD treatment include a blockade of pro-inflammatory cytokines related to Th17 cells. Suppression of IL-17 expression using the oral immunosuppressive drug vidofludimus reduced the proliferation of lymphocytes in vitro. Furthermore, the safety and therapeutic efficacy of vidofludimus was demonstrated in a clinical trial involving IBD patients.88
89 However, IL-17 inhibition is controversial in IBD therapy. Although genetic deletion of the IL-17 receptor improved intestinal inflammation in mice with trinitrobenzenesulfonic acid (TNBS)-induced colitis,90 IL-17A deficiency exacerbated the intestinal inflammatory response in mice with DSS-induced colitis.67 In a clinical trial involving CD patients, secukinumab, a monoclonal antibody that neutralizes IL-17A, showed no therapeutic effect.91 A blockade of both IL-17A and IL-17F led to reduced colonic inflammation in experimental colitis; therefore, this may be a potential strategy for IBD therapy.68
92
2. Inhibition of Specific Transcription Factors Associated with Th17 Cells
Other potential IBD therapies involve blocking the transcription factors associated with Th17 cells. Therefore, these transcription factors have been intensely studied. Transcription factors that are potential pharmacological targets include RAR-related orphan receptor (ROR)γt and STAT3. These transcription factors are essential for the regulation of Th17 proliferation and function. Indeed, one study found that dual inhibition of nuclear factor (NF)-κB and STAT3 attenuated intestinal inflammation.93 Consistent with this, vidofludimus, which can improve experimental colitis by decreasing IL-17A and IL-17F levels via inhibition of NF-κB and STAT3 activation, had a therapeutic effect in IBD clinical trials.88
89 Moreover, pioglitazone, a nuclear receptor peroxisome proliferator-activated receptor γ (PPAR-γ) agonist, decreased Th17 cell differentiation through RORγt and improved DSS-induced colitis.94
95 Recently, inhibition of STAT3 activation also had a positive therapeutic effect. In an experimental DSS-induced colitis mouse model, overexpression of the STAT3 inhibitors metformin and gene-associated retinoid-interferon-induced mortality (GRIM)-19 ameliorated intestinal inflammation and Th17 cell differentiation.4
96
Go to :
ILCs IN IBD
1. Innate Lymphoid Cells
ILCs provide host protective immunity in the mucosal tissues. ILCs are a novel family of effector lymphocytes in IBD that produce IBD-relevant cytokines. ILCs are unique in that they lack antigen-specific receptors and phenotypic markers associated with immune cells but do have a lymphoid morphology.97 The ILC family can be subdivided into 3 subsets based on the types of transcription factors they express for lineage differentiation: ILC1, ILC2, and ILC3. The lineage-specific transcription factors expressed in ILC1, ILC2, and ILC3 are T-bet, GATA-3, and RORγt, respectively.98
99
100 Cytokines secreted by ILCs are the same as those of T effector cells (Fig. 2).
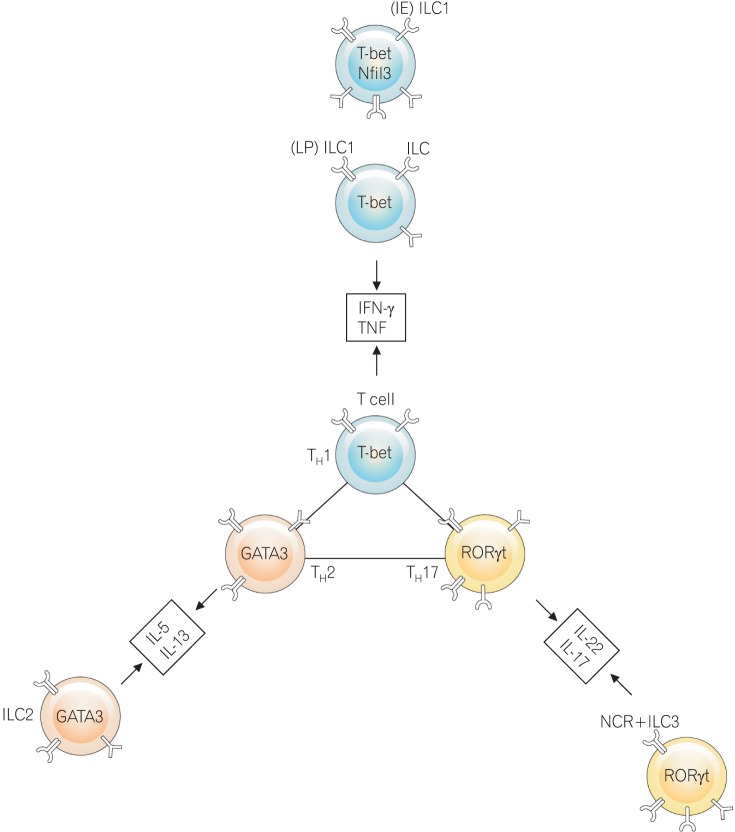 | Fig. 2Pathogenic innate lymphoid cells (ILCs) and T cells in mucosal cells from IBD patients. ILCs have common properties with T effector cells. Lineage-specific transcription factors expressing ILCs or a subset of T cells produce the same cytokine. Natural cytotoxicity receptor (NCR)-expressing ILCs are classified differently from T cells. T-bet, T-box expressed in T cells; IFN, interferon; TNF, tumor necrosis factor; IL, interleukin; RORγt, retinoic acid receptor-related orphan receptor γt.
|
2. ILCs in Intestinal Innate Immunity
ILC3s, in particular, are involved in host defense to extracellular bacteria and fungi.101
102
103 ILC3s can be further subdivided based on whether they express the natural cytotoxicity receptor (NCR). NCR+ILC3 produces IL-22; however, NCR-ILC3, similar to Th17 cells, produces both IL-22 and IL-17.104 On the other hand, RORγt-dependent NCR+ILC3 downregulated RORγt expression and subsequently differentiated into T-bet-dependent IFN-γ-producing cells under IL-12 effects.98
101
105 ILCs that produce IL-17 and IFN-γ are implicated in the pathogenesis of IBD. The pathogenicity of ILC3 was shown in a Helicobacter hepaticus IBD model and a Tbx21−/−
Rag2−/−
UC model.106
107
108
109 Moreover, innate immune cells isolated from IBD patients expressed ILC3 genes (IL17A , IL22, RORC, and IL23R ).110
The levels of T-bet responsive and IFN-γ-producing ILC1 are also higher in CD patients.111
112 IL-12- and IL-15-responsive intraepithelial CD103+NKp46+ILC1 and lamina propria NKp46+ ILC1 were increased in CD patients, and it was suggested that they may have a pathogenic role in the ileum.111
112
113 Meanwhile, ILC2s may contribute to intestinal fibrosis via IL-13 production in the gut. IL-13 producing CD3-KIR+ cells are more abundant in fibrotic areas of the intestine in CD patients.114 Fibrotic lesions have higher levels of IL-13, IL-13Rα2 and collagen expression than non-fibrotic lesions, which is evidence that ILC2s can also aggravate IBD.114
3. Cytokines
Similar to Th17 cells, pathogenic ILC3s are also responsible for IL-23 production, which induces the secretion of IL-17 and IL-22 by ILC3. TNF-α, a key cytokine in IBD pathogenesis, also increased IL-17 production in ILC3s in a mouse model of colitis.107
115 IL-12 stimulates the production of ILC1-specific cytokines in synergy with IL-15 and IL-18.111
112 IL-12 and IL-23 can also contribute to differentiation to either ILC1 or ILC3. It seems that ILC differentiation and contribution to IBD pathogenesis is orchestrated by a combination of these cytokines.111
4. Interaction of ILCs with Mucosal Cells
Interactions between ILCs and immune and non-immune cells determine how ILCs respond to the environment (Fig. 3). Crosstalk between ILCs and mucosal, epithelial, and dendritic cells contributes to the host immune response via ILCs. Mononuclear phagocytes have an important role in the activation of ILCs in the intestine. CD14+CX3CR1+ mononuclear phagocytes produce IL-23, IL-1β, IL-6, TNF-α, and TL1A, which promote the activation of ILCs.116
117
118 CX3CR1+ or CD14+ mononuclear phagocytes mediate ILC3 activation, and this contact is important for ILC3 responsiveness to the gut environment.116
119
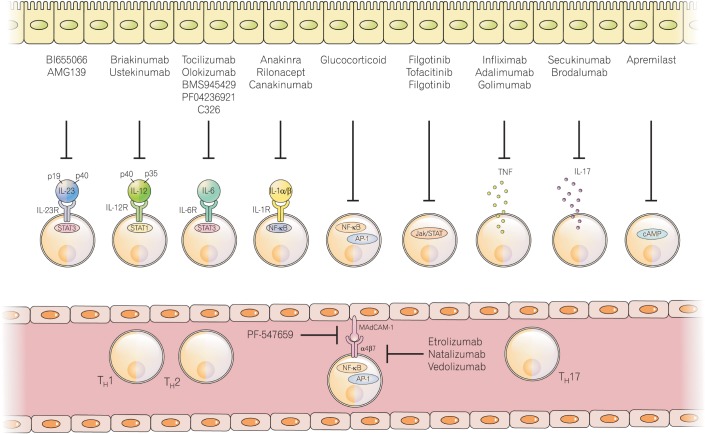 | Fig. 3Present IBD therapeutic strategies that involve prevention of T cell and innate lymphoid cells (ILC) production or their inhibition. T cells and ILCs have a common therapeutic target. Compared with classical IBD therapeutic agents, new therapeutic strategies may involve T cells; ILCs such as interleukin (IL)-23 and IL-12-, tumor necrosis factor (TNF)-, and integrin-targeting agents; and signal transducer and activator of transcription (STAT) inhibitors. NF, nuclear factor; AP-1, activator protein 1; cAMP, cyclic adenosine 3′:5′-monophosphate.
|
ILCs also interact with Treg cells, which are important for intestinal immune control. Commensal bacteria-responsive, IL-1β-producing mononuclear phagocytes induce GM-CSG secretion by ILC3s, and these ILC3s produce retinoic acid and TGF-β for Treg cell differentiation.120
With the exception of ILC1, ILC2 and ILC3 express major histocompatibility complex (MHC) class II and can influence CD4+T cells. ILC2 activates Th2 cell differentiation through MHC class II, CD80 and CD86.121 ILC3 that is lacking CD80, CD86, and CD40 cause dysregulated T-cell regulation and increased IL-17 secretion, illustrating the immunoregulatory role of ILC3 in gut T cells.122
123
124 Interactions between ILCs and B cells promote Ig production T-cells-independently. Thus, B-cell activating factor (BAFF), CD40L and Notch ligand delta-like 1 (DLL1) are increased by ILCs interaction in splenic marginal zone and augments antibody secretion by B1 cells.125 ILC3s also produce IL-10 and express the CCL60 receptor, CCR6, for trafficking to Peyer's patches and the intestinal epithelium. These properties of ILC3 are dependent on IL-22 signaling, because a lack of IL-22 causes a loss in tolerance to commensal bacteria and unchecked growth of pathogenic bacteria, which, together, increase the probability of developing colitis.126
127
128
Although cytokines secreted by ILCs are very similar to T cells, this new population of cytokines has unique property that expresses both receptors for T cells and NK cells. In IBD patients, ILCs are abundant in inflamed lesions of the intestine; therefore, ILCs have a pathological role and should be considered targets in the development of future IBD therapies. Moreover, ILCs mediate environmental signals for T and B cell development. Therefore, ILCs should be validated in IBD patients and may be key targets to treat IBD.
Go to :
IBD THERAPEUTICS
1. Classical Drugs for IBD Treatment
There are 2 main categories of therapeutics for treating IBD: (1) anti-inflammatories or immunosuppressive agents, and (2) biological agents. Classically, anti-inflammatory drugs, such as 5-aminosalicylates (5-ASAs), are used to treat UC. 5-ASAs are effective for maintenance of remission in UC and also reduce tumor development by inhibiting PG synthesis, reducing pro-inflammatory cytokine levels, blocking neutrophil attraction and activating mast cells by inhibiting NF-κB in immune cells, and promoting PPAR-γ expression and nuclear translocation.129
130
131
132 Unfortunately, 5-ASAs have little efficacy in CD patients.133 Corticosteroids are also used for induction of remission in UC patients. Glucocorticoids inactivate NF-κB, activator protein 1 and prevent the production of inflammatory cytokines such as IL-1 and IL-6.134
135
136
Azathioprine, methotrexate, and cyclosporine-A are classical immunosuppressive drugs used in IBD therapy. Methotrexate is used for maintenance of remission in CD and cyclosporine-A is used to induce remission state of UC, while azathioprine has efficacy in both CD and UC.137
138
139
140
141 Cyclosporine-A and methotrexate suppress the secretion of pro-inflammatory cytokines and induce apoptosis.142
143
144
145 Cyclosporine-A suppresses the production of IL-22 and TNF-α by NCR+ILC3.146
TNF-α is the main pathogenic factor that is produced by immune and non-immune cells in the gut of IBD patients. Anti-TNF agents, including infliximab, adalimumab, and golimumab, are classic IBD therapies. Combination therapy with infliximab and azathioprine is very effective for maintenance of remission in both CD and UC.139
147
148 However, IBD treatments that involve simply blocking or neutralizing the TNF receptor using incomplete antibodies, such as etanercept, are not effective because such antibodies have a short half-life and low efficacy, consistent with the in vivo results of anti-TNF therapy.149
150
2. Inhibition of Lymphoid Cell Homing
In the last decade, clinical trials have shown that 30% to 50% of IBD patients do not respond to anti-TNF therapy. Therefore, various strategies are attempted to target IBD mechanism. Natalizumab, an antibody targeting α4 integrin, is a promising new target for CD therapy and works by blocking T-cell recruitment into lesions via α4β7 integrins, 151
152
153
154 however, risk of progressive multifocal leukoencephalopathy in the use of natalizumab was reported.155
156 Some ILC subsets also express α4β7 integrins for homing to the gut.98
157 Thus, the more specific α4β7 integrin blocker, vedolizumab, was developed and showed good efficacy in both CD and UC.158
159
160
161
162 Vedolizumab also can be used in patients who do not respond to anti-TNF agents.163 Starting with anti-α4β7 therapy, subsequent trials involved the inhibition of T-cell homing using antibodies against MAdCM1 and β7 integrin.164
165
166
3. Inhibition of IBD-Related Lymphoid Cell Survival
Some IBD therapies target signaling pathways specific to lymphoid cell survival. Sphinogosine-1-phosphate (S1P) and G protein-coupled receptor (S1PR) signaling pathways both activate NF-κB and STAT3, resulting in T-cell proliferation and angiogenesis.167 In clinical trials, ozanimod, a S1PR agonist, induced lymphopenia, suppressed experimental colitis, and induced remission of UC.168
169
170 Preclinical challenges for pathogenic ILCs have been developed too. In a mouse colitis model, the ILC-specific cytokine, IL-7, controlled ILC3 survival and ILC3-specific cytokine production. Additionally, blockade of IL-7R effectively reduced intestinal ILCs.107
146 In other studies, CD90 inhibition depleted ILCs and ameliorated IBD in a mouse model.106
158
4. Targeting Epithelial Cells
Intestinal epithelial cells can also be targeted to treat IBD. MMP9 (GS-5746) is an enzyme that induces proteolysis, stimulates the infiltration of immune cells into inflamed gut tissue, and increases TNF-α levels. In a clinical trial, however, targeting MMP9 failed to reduce inflammation in UC patients.171 Repeated cycles of inflammation and healing also induce the formation of tissue fibrosis. Small-interfering RNAs mediated silencing of CHST15, a chondroitin synthetic enzyme, resulted in reduced α-SMA levels in fibroblasts and reduced collagen deposition in animal models of colitis. CHST15-specific small-interfering RNA (STNM01) reduced inflammation and fibrosis in CD clinical trials.172
5. Targeting Cytokines in IBD Therapy
Main cytokines related with IBD were classical targets for IBD treatment. Surprisingly, studies showed that anti-IFN-γ and anti-IL-17A antibody therapies aggravated CD, possibly due to unexpected effects or their protective roles in gut epithelial cells.91
173
174 IL-6 is one of the main pro-inflammatory cytokines that activates immune cells. The IL-6-targeting antibody tocilizumab (previously known as MRA) showed high efficacy in a colitis model and induced remission in CD patients.175
176 Considering the role played by IL-6 signaling in the proliferation of gut epithelial cells, the therapeutic value of anti-IL-6 or anti-gp130 (IL-6R) antibodies should be investigated. The pro-inflammatory cytokines IL-23 (heterodimer of p19 and p40) and IL-12 (heterodimer of p19 and p40) cause inflammation by induction of Th17, Th1 or ILCs development in the gut mucosa of CD patients.177
178 Therefore, anti-p40 and p19 antibodies targeting IL-23 and IL-12 have been developed. In clinical trials involving p40 blockers, such as ABT-874 and ustekinumab, a high response was induced in CD patients, even among patients who had anti-TNF therapy.179
180 Neutralization of IL-23 (p19) also blocked the stimulation of pathogenic ILC3s and the production of IL-17A- and IL-22-producing cells.106
107 Clinical trials are currently investigating the efficacy of p19-targeting drugs (BI655066 and AMG139) in CD patients.181 Other targets in IBD therapy include cytokine-related signaling proteins, including Janus kinases (JAK1-3 and TYK2), which can suppress the secretion of cytokines into the mucosa. In another study, the JAK inhibitor tofacitinib was promising in UC but not CD patients.182
183 In contrast, the JAK1-specific inhibitor filgotinib stabilized the remission state in CD patients. Phos phodiesterase-4 inhibition by apremilast negatively regulated cyclic adenosine 3′5′-monophosphate (cAMP), a key mediator of inflammation, by suppressing the pro-inflammatory cytokines IFN-γ, TNF-α, IL-12, IL-17, and IL-23.184
Go to :
CONCLUSIONS
Although the pathogenesis of IBD is complicated and involves many pro-inflammatory mediators, it is clear that Th17 cells play a central role in the induction and maintenance of chronic intestinal inflammation in IBD patients. With regard to reducing intestinal inflammation, research has shown that Th17 cells should be the primary targets for IBD therapy. Many studies have shown that Th17 cells have a pathogenic roles in intestinal inflammation; however, there is still much to be investigated. Therefore, a better understanding of Th17 cells and their targeting could lead to the development of an effective IBD therapy. ILCs should be considered for IBD therapy. ILCs have common therapeutic targets with Th17 cells and are abundant in the gut of IBD patients. Further studies on the role of ILCs in gut immunity would lead to the development of better IBD therapies (Fig. 4).
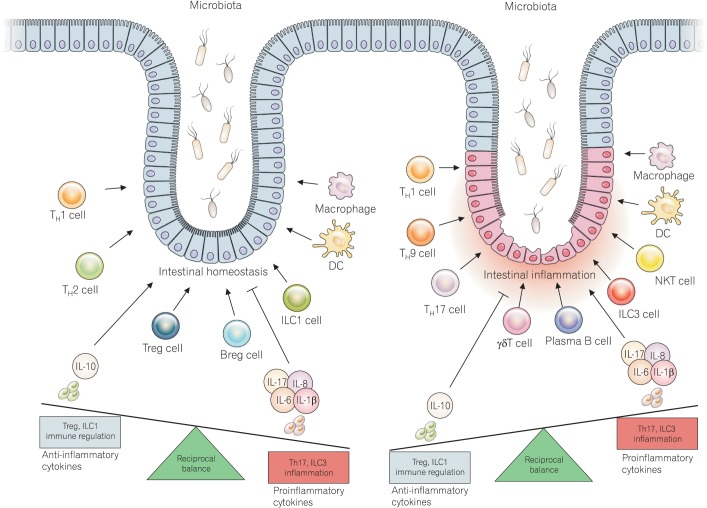 | Fig. 4Reciprocal balance for intestinal immune homeostasis and inflammation. The normal state is mediated by a reciprocal balance between immune cells (Treg and Breg vs. Th17 and ILC1) and cytokines that are secreted to maintain the conditions in the intestine. However, an imbalance in immune cells leads to the destruction of intestinal epithelial cells and the invasion of commensal microbiota. This situation leads to the uncontrolled release of cytokines, which is a key event in the pathogenesis of IBD. Treg, regulatory T; Breg, regulatory B; ILC, innate lymphoid cells; DC, dendritic cell; IL, interleukin; NKT, natural killer T; Th17, T helper 17.
|
Beyond innate immunity, adaptive immunity also has a direct role in the pathogenesis of IBD. An overwhelming number of effector cells, such as Th17 cells and ILCs, induce self-destructive immunity; therefore, a cure for IBD would involve understanding how immunological balance is controlled.
Go to :
 XML Download
XML Download