

 PDF
PDF Citation
Citation Print
Print



INTRODUCTION
Autoimmune enteropathy (AIE) is a rare disease characterized by intractable diarrhea and the presence of anti-enterocyte autoantibodies [1]. Most cases of AIE harbor mutations in FOXP3, a master gene of regulatory T cells (Tregs), and are caused by immune dysregulation, polyendocrinopathy, enteropathy, X-linked (IPEX) syndrome, which is characterized by multi-organ autoimmunity [2,3]. The AIE-related 75 kDa antigen (AIE-75) and villin have been established as target antigens recognized by anti-enterocyte autoantibodies and detected in the sera of patients with IPEX syndrome [4,5]. Although intractable enteropathy is also associated with mutations in other Treg-associated genes such as CD25 and CTLA4, enterocyte antigens have not been identified [6-8]. We report a case of a 13-year-old girl with CTLA4 haploinsufficiency, who was positive for anti-AIE-75 autoantibody and was successfully treated with 6-mercaptopurine (6-MP). The written informed consent was waived by the ethics committee of our hospital.
CASE REPORT
A 13-year-old girl was admitted to our hospital mainly complaining of persistent watery diarrhea. She was diagnosed with immunoglobulin A (IgA) deficiency with low IgG2 and IgG4 subclass, and had a history of 4 previous admissions since age 11 because of recurrent immune thrombocytopenic purpura (ITP), sometimes accompanied by autoimmune hemolytic anemia, leukopenia, and cold agglutinin disease. She was also suspected of having interstitial nephritis. She responded well to prednisolone (PSL; 2 mg/kg/day) that was used to treat ITP at every onset, and the treatment was successfully tapered off. However, watery diarrhea appeared during the tapering of PSL while the treatment for the fifth episode of ITP was ongoing. She had no family history of autoimmune diseases, except for hypothyroidism in her mother and hyperthyroidism in her sister.
Her height was 155 cm (third to fifth percentile) and weight was 48 kg (fifth percentile). On physical examination, her body temperature was 36.0°C with no particular abnormal findings including exanthema. Laboratory examination revealed the white blood cell count to be 13,300/μL; platelet count, 21.8 × 104/µL; hemoglobin, 13.7 g/dL; aspartate aminotransferase, 14 U/L; alanine aminotransferase, 13 U/L; lactate dehydrogenase, 236 U/L; blood urea nitrogen, 11.1 mg/dL; creatinine, 1.6 mg/dL; sodium, 135 mEq/L; potassium, 3.2 mEq/L; chloride, 112 mEq/L; total protein, 6.3 g/dL; albumin, 4.2 g/dL; C-reactive protein, 0.13 mg/dL; IgG, 476 mg/dL; IgA, < 4 mg/dL; IgM, 51 mg/dL; and soluble interleukin-2 receptor (sIL-2R), 3,280 U/mL. None of the viruses, namely, adenovirus, enterovirus, cytomegalovirus, Epstein-Barr virus, norovirus, and rotavirus, were detected in the patient’s stool sample using polymerase chain reaction. In addition, no pathogenic bacteria were cultured from the stool. Besides, a toxin detection test for Clostridium difficile also yielded negative results. A computed tomography scan of her abdomen revealed a small volume of ascites.
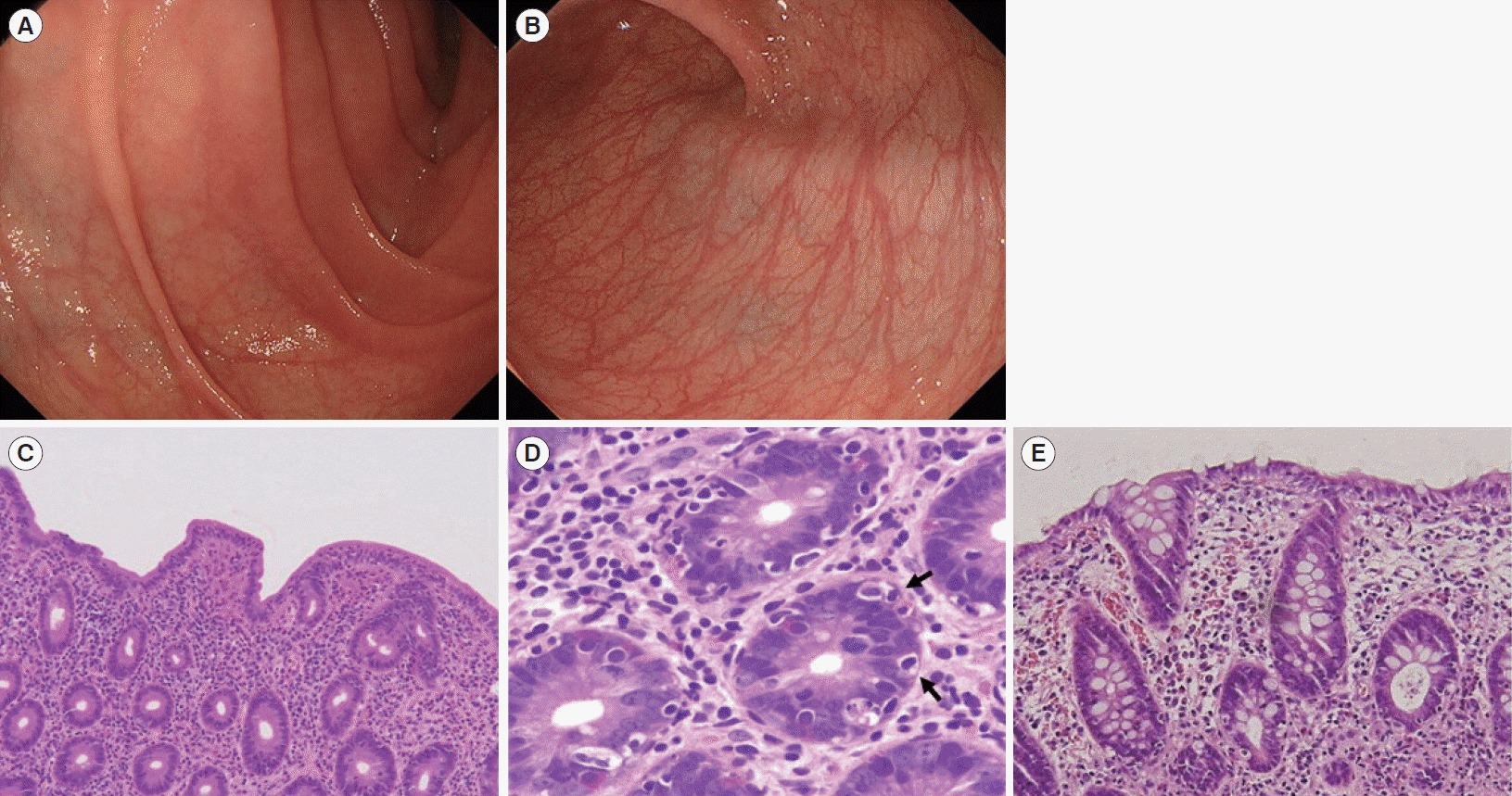
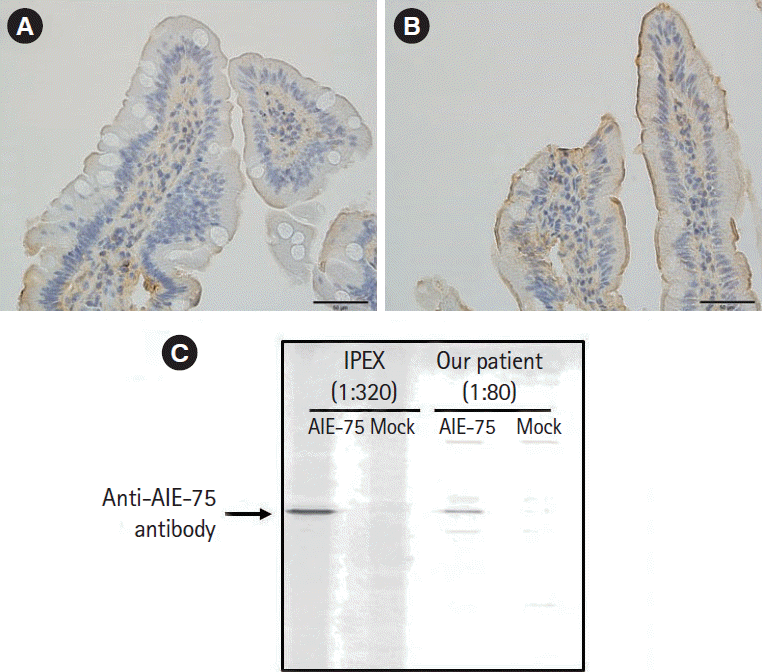
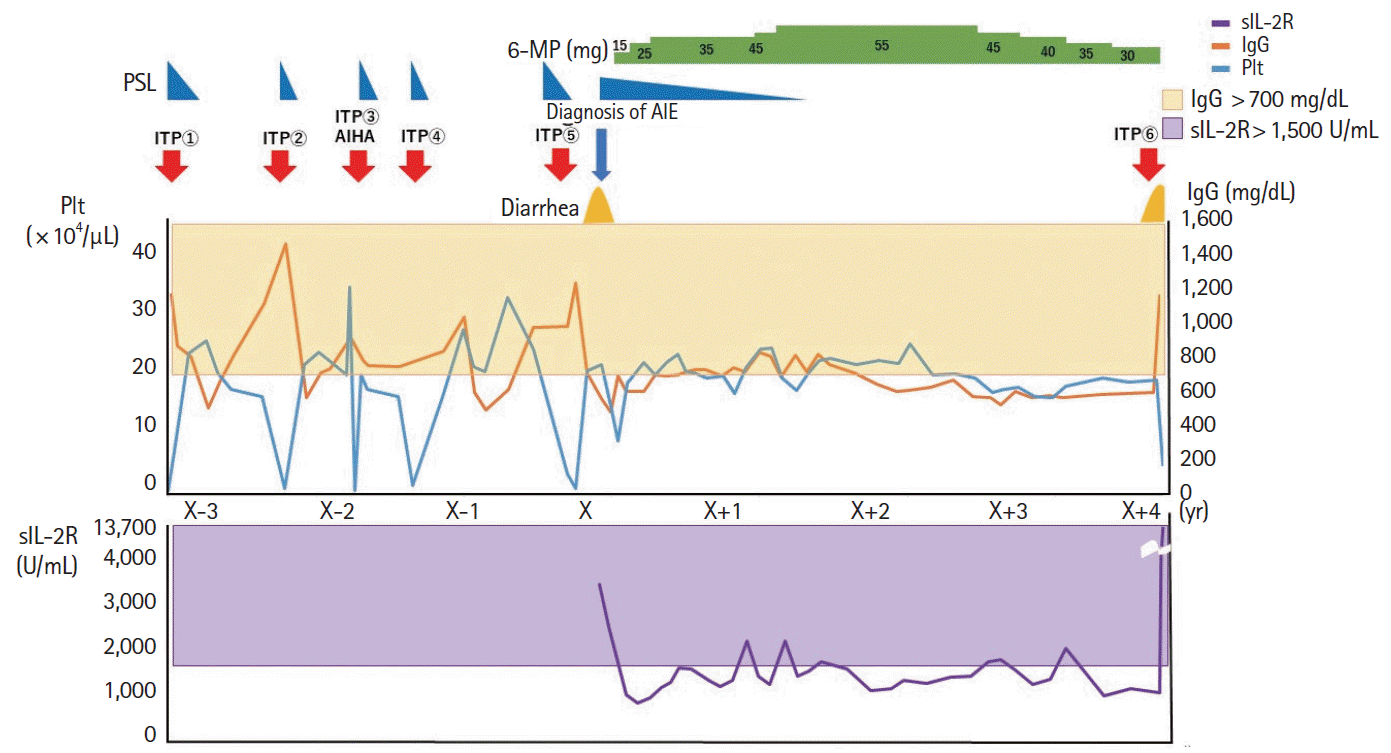
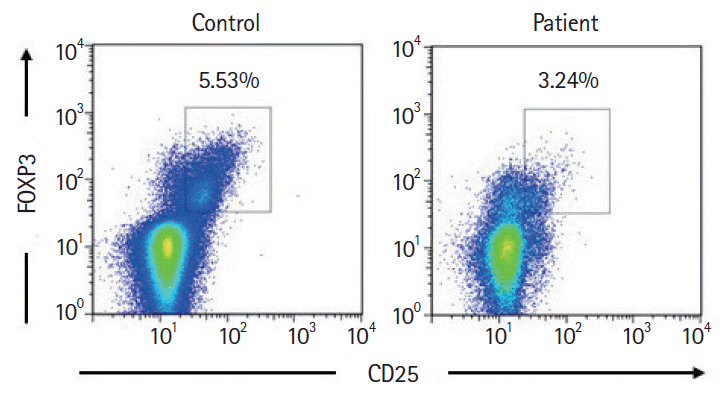
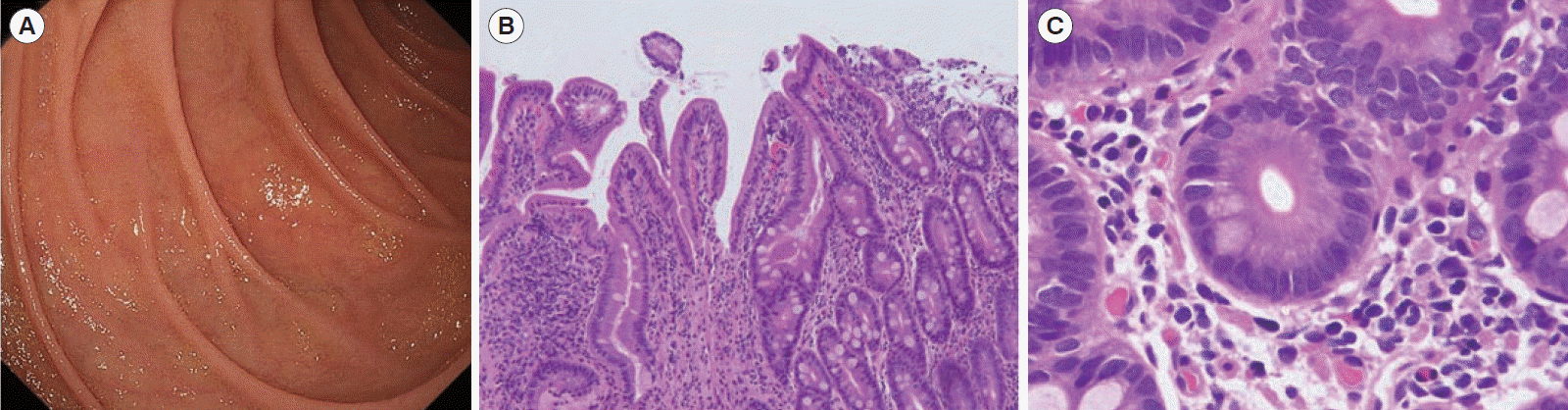
Diarrhea persisted despite dietary restrictions with total parenteral nutrition, administration of probiotics and anti-diarrheal drugs, or cessation of a proton pump inhibitor. An esophagogastroduodenoscopy and a colonoscopy demonstrated prominent atrophy of the villus in the duodenum (Fig. 1A) and terminal ileum. Colon was intact endoscopically (Fig. 1B). Histopathological examinations of the duodenum and ileum showed severe villous atrophy. Crypt apoptosis, and infiltration of lymphoplasmacytes into the lamina propria were seen in the biopsies of duodenum, ileum, and colon (Fig. 1C-E). Immunostaining of the normal small intestine tissue with the patient’s serum showed positive staining of the brush borders of the intestinal epithelium, indicating the presence of anti-enterocyte autoantibodies in the serum of the patient (Fig. 2A and B). Furthermore, autoantibodies against AIE-75 were detected by Western blotting, using a recombinant protein as an antigen (Fig. 2C) [4]. The patient was diagnosed with AIE and was administered an increased dose of PSL (40 mg/day) as induction therapy. Because a previous report of 14 cases of pediatric AIE showed that some patients were treated with immunomodulators as a maintenance therapy with a partial to complete response [9], we decide to introduce 6-MP in the same manner as the treatment of inflammatory bowel disease for its steroid-sparing effect in this patient. After analyzing the patient’s past laboratory data, we found that following cessation of PSL, ITP relapsed in association with the normalization of IgG levels (861–1,747 mg/dL). In addition, we found that serum levels of sIL-2R were extremely elevated at the onset of AIE. There was no manifestation of AIE, ITP, or other autoimmune diseases for 4 years, while the dose of 6-MP was adjusted to keep the levels of IgG and sIL-2R below 700 mg/dL and 1,500 U/mL, respectively (Fig. 3). Whole-exome sequencing demonstrated a heterozygous missense mutation in the CTLA4 gene. Flow cytometry analysis showed reduced frequency of CD4+ CD25+ FOXP3+ Tregs (Fig. 4) [10]. Consistent with clinical remission of AIE, an endoscopy and histological study 3 years after the achievement of remission demonstrated recovery from villous atrophy (Fig. 5). After 4 years of 6-MP administration, we tried to reduce the amount of 6-MP because we were concerning about the risk of secondary malignancies such as malignant lymphoma caused by 6-MP. Unfortunately, it led to the relapse of diarrhea and ITP along with soar up of serum level of rIL-2 receptor (Fig. 3).
DISCUSSION
We report a patient with intractable diarrhea, carrying a heterozygous substitution of c.119T > C (p.Val40Ala) in exon 2 of the CTLA4 gene, which has been included as P72 (Family BB) in a large international cohort study [11]. Valine 40 is located in the ligand-binding domain and is highly conserved between different species. Although the pathological role of the amino acid substitution remains unclear, the patient’s clinical features suggest that the substitution is a mutation rather than a polymorphism, and the diagnosis is that of CTLA4 haploinsufficiency. The patient’s mother and sister were found to have the same heterozygous mutation, but they showed only thyroid gland dysfunction that was manageable with medication, which suggests incomplete penetrance. The clinical penetrance of the disease is reportedly estimated to be 67% [11]. In the international cohort study about CTLA4 mutation carriers, intractable diarrhea are observed in as much as 57% [11]. It does not clearly mention that those diarrhea are attributed to AIE, therefore, the exact incidence rate of AIE in patients with CTLA4 haploinsufficiency is unclear. Given that a case report of CTLA4 haploinsufficiency included the information of the presence of AIE [6,12], together with our present case, at least some parts of 57% patients are presumed to meet the criteria of AIE.
Celiac disease is also reported as a complication of CTLA4 haploinsufficiency [11]. Although celiac disease was not excluded by the histopathological findings in the small intestine, unresponsiveness to dietary restrictions suggests AIE rather than celiac disease [1,13]. Furthermore, we detected anti-enterocyte autoantibodies, thereby confirming the diagnosis of AIE. The original diagnostic criteria for AIE consisted of (1) severe and protracted diarrhea, (2) lack of response to dietary restrictions, (3) presence of circulating gut antibodies and/or associated autoimmune disease, and (4) lack of severe immunodeficiency [1]. The last item of the criteria (lack of severe immunodeficiency) may indicate that most cases of AIE are caused by IPEX syndrome [9].
Notably, anti-AIE-75 antibodies were detected in the sera of our patient. CTLA4 is expressed on activated effector T cells and attenuates their activation [14]. Because CTLA4 is also constitutively expressed in Tregs, which compete with CD28 on effector T cells for binding CD80/86 and possibly downregulate the expression of CD80/86 molecules, defects of CTLA4 lead to impaired Treg function despite a normal or subnormal number of Foxp3-positive T cells [15]. Thus, it is not surprising that the same autoantibodies are detected in both IPEX syndrome and IPEX-like syndrome. Since AIE-75 is also expressed in the brush border of proximal renal tubules [4], antigen-specific immunity may be involved in the development of interstitial nephritis in our patient. Chida et al. [16] have reported that antiAIE autoantibodies were detected in patients with IPEX, but not in patients with autoimmune polyendocrinopathy, candidiasis, ectodermal dystrophy (APECED). Instead, anti-tryptophan hydroxylase-1 antibodies are specific to APECED with intestinal disorders [16]. Given that APECED is caused by the deficiency of the autoimmune regulator gene, which is critical for central tolerance, immunotolerance to AIE-75 may depend on the peripheral mechanism. Our present results support this idea.
Ipilimumab, a monoclonal antibody against CTLA4, can induce immune-related adverse events. The incidence of gastrointestinal adverse event by ipilimumab is reported as 35% [17]. It is assumed that depletion of Treg and activation of effector T cells in the gastrointestinal tract lead to colitis [18]. The mechanism of colitis onset by ipilimumab seems to be shared with mechanism of AIE. Further investigation on the relation between digestive symptoms and CTLA4 dysfunction are warranted.
In our patient, clinical remission was achieved with 40 mg/day of PSL. Clinical remission was maintained on 6-MP even after cessation of PSL for 4 years. Since CTLA4 forms a homodimer [14], one-fourth of the expressed molecules are theoretically functional in haploinsufficiency. Thus, the Treg function may be partially impaired in CTLA4 haploinsufficiency, while it is almost completely impaired in IPEX syndrome. This is consistent with the late onset of autoimmune diseases in CTLA4 haploinsufficiency than in IPEX syndrome, where multi-organ autoimmunity, including AIE, develops during early infancy [3,11]. In the case of inflammatory bowel diseases, such as ulcerative colitis and Crohn’s disease, the optimal dose of 6-MP is monitored using the white blood cell count and 6-thioguanine nucleotide level [19,20]. In contrast, we found that elevated serum levels of both IgG (> 700 mg/dL) and sIL-2R (> 1,500 U/mL) are associated with relapse of ITP in our patient. Furthermore, AIE was controlled by keeping both markers below those levels, without obvious adverse events. These findings suggest that both humoral and cellular immunity play critical roles in the development of AIE. Indeed, CD4+ T cells are major infiltrating cells in the enteropathy associated with CTLA4 haploinsufficiency [6,7], although the subsets of infiltrating lymphocytes in the intestine were not tested in our patient.
In conclusion, anti-AIE-75 autoantibodies were detected in a case of AIE associated with CTLA4 insufficiency, and the use of 6-MP following induction therapy with glucocorticoids was effective for both ITP and AIE. The serum levels of sIL-2R and IgG could be used for monitoring 6-MP therapy.
 XML Download
XML Download