

 PDF
PDF Citation
Citation Print
Print



INTRODUCTION
Ulcerative colitis (UC) is a chronic, relapsing, inflammatory disorder of the large bowel that is characterized by a lymphocytic infiltration of the colonic mucosa with resulting loss of mucosal architecture [1]. UC requires long-term management for the induction and maintenance of clinical remission, and adherence to medication is an important challenge [2].
Subcutaneous (SC) administration of biologics is effective, safe, and well-tolerated. In general, SC is preferred by both patients and healthcare providers, and can be self-administered at home [3]. Currently, 3 of the 5 licensed biologics that are approved for the treatment of inflammatory bowel disease (IBD) in Japan use the SC route of administration [4-8]. The tumor necrosis factor (TNF)-α antagonists adalimumab and golimumab are approved for SC administration for the treatment of UC [6,7]. The interleukin-12/23 p40 antagonist ustekinumab is approved for SC administration as maintenance therapy for refractory moderate to severe UC patients [8].
Vedolizumab is a humanized, gut-selective immunoglobulin G1 monoclonal antibody that binds to α4β7 integrin and blocks lymphocyte infiltration to the gut tissue [9,10]. Vedolizumab administered through intravenous (IV) infusion (vedolizumab IV) is approved in Japan to treat patients with moderate to severe UC or Crohn’s disease (CD) [4]. The efficacy and safety of vedolizumab has been evaluated in randomized, placebo-controlled, phase 3 trials in Japan of patients with UC (n = 292) [11] or CD (n = 157) [12]. Results from both trials showed that vedolizumab IV had: numerically, but not significantly, greater efficacy over placebo as induction therapy (at week 10); was either numerically or significantly superior over placebo as maintenance therapy (at week 60); and was well tolerated in Japanese patients with UC [11] or CD [12]. In contrast, results from the global, pivotal, phase 3 GEMINI 1 study in a larger number of patients with UC (n = 895) demonstrated that vedolizumab IV had significantly greater efficacy over placebo as induction therapy (at week 6: all outcome measures P<0.001 or P=0.001) and maintenance therapy (at week 52: all outcome measures P<0.001 or P=0.001) [13]. Other differences in clinical characteristics have been reported between patients with IBD from Japan and those from other countries. For example, a recent administrative database analysis of Japanese patients with IBD found no increase in the incidence of non-Hodgkin lymphoma attributable to thiopurine treatment [14]. However, cohort studies in European patients with IBD reported an association between thiopurine treatment and an increased risk of lymphoproliferative disorders or cancer [15,16]. It remains to be determined whether the efficacy and safety of vedolizumab SC differs between patients with UC from Japanese and global populations.
The efficacy and safety profile of vedolizumab SC as maintenance therapy was evaluated in the phase 3 VISIBLE 1 study of patients with moderate to severe UC who achieved clinical response at week 6 following vedolizumab IV induction therapy (ClinicalTrials.gov NCT02611830; EudraCT 2015-000480-14) [17]. Here, we report results from the Japanese subgroup of patients with UC who were enrolled in VISIBLE 1.
METHODS
1. Study Design, Randomization, and Blinding
This was a subgroup analysis of the Japanese patients who were enrolled in the VISIBLE 1 study, which was a pivotal, phase 3, multicenter, multinational, randomized, double-blind, double-dummy, placebo-controlled trial [17]. This subgroup analysis evaluated the efficacy and safety of maintenance treatment with vedolizumab SC in adult patients with moderate to severe UC who achieved a clinical response at week 6 with open-label therapy with vedolizumab IV 300 mg at weeks 0 and 2. A vedolizumab IV reference arm was included for descriptive comparisons of efficacy, safety, and immunogenicity with vedolizumab SC.
In the induction phase, eligible patients were enrolled at week 0, received open-label infusions of vedolizumab IV 300 mg at weeks 0 and 2, and were assessed for clinical response by complete Mayo score at week 6 (defined as a reduction in complete Mayo score ≥ 3 points and ≥ 30% from week 0, with an accompanying decrease in rectal bleeding subscore ≥ 1 point or absolute rectal bleeding subscore ≤ 1 point). Patients with clinical response entered the maintenance phase and those with no clinical response received a third IV infusion of vedolizumab 300 mg at week 6. Patients with clinical response by partial Mayo score at week 14 (defined as a reduction in partial Mayo score ≥ 2 points and ≥ 25% from week 0, with an accompanying decrease in rectal bleeding subscore ≥ 1 point or absolute rectal bleeding subscore ≤ 1 point) were eligible to enroll in an open-label extension study (NCT02620046; EudraCT 2015-000482-31). Patients with no clinical response were discontinued.
Patients with clinical response at week 6 were enrolled in the maintenance phase. Patients were randomized in a double-blind, double-dummy fashion to receive SC injections every 2 weeks (Q2W) and IV infusions every 8 weeks (Q8W), from week 6 through week 50, as follows: vedolizumab SC injections 108 mg Q2W and placebo IV infusions Q8W; or placebo SC injections Q2W and placebo IV infusions Q8W; or vedolizumab IV infusions 300 mg Q8W and placebo SC injections Q2W. SC prefilled syringes were used. Further details on SC injection technique are provided in the Supplementary Material.
Randomization was stratified by concomitant use of oral corticosteroids, clinical remission status at week 6, and previous failure with TNF-α antagonist treatments or use of a concomitant immunomodulator (azathioprine or 6-mercaptopurine).
Further details on randomization and blinding are provided in the Supplementary Material.
2. Patients
Eligible patients were aged 18–80 years and were diagnosed with UC ≥ 6 months before study enrollment. Patients had moderate to severe UC, defined as complete Mayo score 6–12 with an endoscopic subscore ≥ 2; and had inadequate response to, loss of response to, or intolerance to immunomodulators (azathioprine or 6 mercaptopurine), TNF-α antagonists, or corticosteroids. At week 6, patients who received oral corticosteroids and achieved clinical response began a corticosteroid tapering regimen (further details in Supplementary Material). The institutional review board of each study site reviewed and approved the study protocol. This study was conducted in compliance with all institutional review board regulations and in accordance with the ethical principles that have their origin in the Declaration of Helsinki. Informed consent was provided by all patients. For patients aged < 20 years, additional consent from parents or guardians was required.
3. Endpoints
1) Efficacy
Efficacy analyses for the induction phase (week 6) were conducted in patients enrolled at week 0. Efficacy analyses for the maintenance phase (week 52) were conducted in patients enrolled at week 0 who had clinical response at week 6.
The primary efficacy endpoint was clinical remission (defined as complete Mayo score ≤ 2 points and no individual subscore > 1 point) at week 52. Secondary efficacy endpoints included: endoscopic improvement (referred to as mucosal healing in the study protocol) at week 52, defined as Mayo endoscopic subscore of ≤ 1 point; durable clinical response (defined as clinical response at week 6 and week 52); durable clinical remission (defined as clinical remission at week 6 and week 52); and corticosteroid-free remission (defined as discontinuation of oral corticosteroids, with subsequent clinical remission at week 52, in patients receiving oral corticosteroids at baseline). Clinical remission at week 52 by prior TNF-α antagonist therapy was an exploratory endpoint. Further details on assessments are provided in the Supplementary Material.
2) Patient-Reported Outcomes
Inflammatory Bowel Disease Questionnaire (IBDQ) total score and subscores, Euro Quality of Life-5DE (Q-5D) visual analog scale (VAS) score, and Work Productivity and Activity Impairment (WPAI-UC) instrument scores were assessed. Further details on patient-reported outcomes (PROs) are provided in the Supplementary Material.
3) Safety/Tolerability
Adverse events (AEs), defined as any AE regardless of relationship to study drug, were captured during study visits and from any spontaneous reports at any time during the study, and were coded using the Medical Dictionary for Regulatory Activities (MedDRA; version 21.0).
4) Immunogenicity
The rates of anti-vedolizumab antibody (AVA) development were evaluated using a validated electrochemiluminescence assay [18]. Positive AVA status was defined as ≥ 1 positive AVA result from predose to week 52. Persistently positive AVA status was defined as having a serum sample positive for AVA at ≥ 2 consecutive visits.
5) Statistical Analysis
This is an ad-hoc analysis of the Japanese subgroup of patients with UC enrolled in the VISIBLE 1 study. The majority of statistical methods are the same as those described in the VISIBLE 1 study [17]. However, statistical tests were not performed owing to the lower number of Japanese patients with UC in this ad-hoc analysis.
RESULTS
1. Patient Disposition and Baseline Characteristics
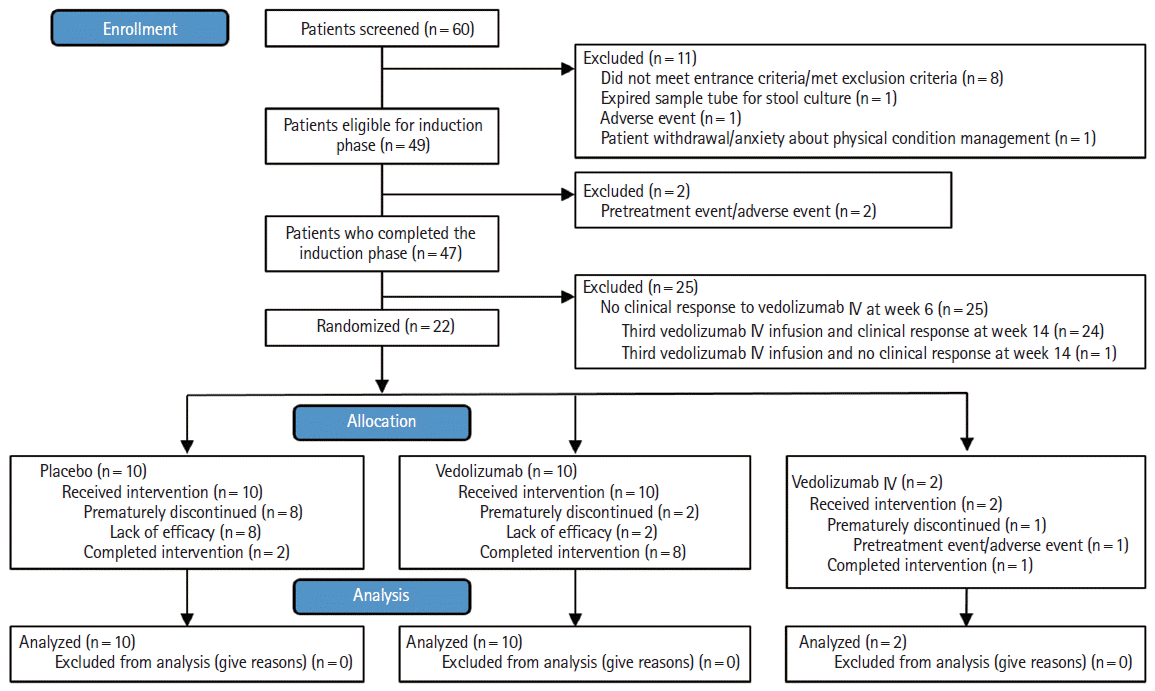
Demographic and baseline characteristics, including racial background, of patients enrolled in the VISIBLE 1 study are shown in Supplementary Table 1. Of the 60 patients screened across 24 sites in Japan, 49 were eligible to enter the induction phase from April 2016 to April 2017 (Fig. 1). At week 6, 22 out of 49 patients (45%) had a clinical response and were eligible for randomization to receive the following treatments in the maintenance phase: vedolizumab SC 108 mg (n = 10), placebo (n = 10), or vedolizumab IV 300 mg (n = 2).
Baseline characteristics for patients enrolled in the maintenance phase were generally similar to those for patients in the induction phase (Table 1). The mean duration of UC was 7.1 ± 4.0 years in the vedolizumab SC group, 10.3 ± 6.6 years in the placebo group, and 8.2 ± 8.0 years in the vedolizumab IV group (Table 1). Prior failure of TNF-α antagonist therapy had occurred in 5 out of 10 patients (50%) in the vedolizumab SC group, 2 out of 10 patients (20%) in the placebo group, and 2 out of 2 patients (100%) in the vedolizumab IV group.
2. Efficacy
A numerically higher rate of clinical remission was observed at week 52 in the vedolizumab SC group (4/10, 40%) than in the placebo group (2/10, 20%) (difference between groups, 20% [95% confidence interavl (CI), –27.9 to 61.8]) (Table 2). In the vedolizumab IV group, 1 out of 2 patients (50%) had clinical remission at week 52.
The same rates and trend were observed for the proportion of patients with endoscopic improvement at week 52 in all treatment groups (vedolizumab SC group, 40%; placebo group, 20%; and vedolizumab IV group, 50%) (Table 2). A higher proportion of patients with durable clinical response was observed at week 52 in the vedolizumab SC group (8/10, 80%) than the placebo group (2/10, 20%) (difference between groups, 60.0% [95% CI, 12.7 to 88.5]) (Table 2). In the vedolizumab IV group, 1 out of 2 patients (50%) had durable clinical response at week 52. A numerically greater proportion of patients with durable clinical remission was observed at week 52 in the vedolizumab SC group (2/10, 20%) than the placebo group (1/10, 10%) (Table 2). Corticosteroid-free remission at week 52 was observed in 1 out of 4 (25%) of patients in the vedolizumab SC group and 1 out of 3 (33%) of patients in the placebo group (Table 2). No patients in the vedolizumab IV group had durable clinical remission at week 52.
In patients who failed prior TNF-α antagonist therapy, clinical remission at week 52 was observed in 1 out of 5 patients (20%) in the vedolizumab SC group and in 0 out of 2 patients in the placebo group (difference between groups, 20.0% [95% CI, –65.4 to 84.1]) (Table 3). In patients who were TNF-α antagonist therapy naïve, clinical remission at week 52 was observed in 3 out of 5 patients (60%) in the vedolizumab SC group and 2 out of 8 patients (25%) in the placebo group (difference between groups, 35.0% [95% CI, –23.4 to 78.9]) (Table 3).
A summary of fecal calprotectin levels by study visit is shown in Supplementary Table 2.
3. Patient-Reported Outcomes
In all patients, IBDQ and EQ-5D VAS PRO instrument scores increased by week 6 following open-label, IV induction therapy with vedolizumab at weeks 0 and 2 (Supplementary Figs 1, 2). During maintenance treatment, IBDQ and EQ-5D VAS scores generally decreased for patients receiving placebo, while patients receiving vedolizumab SC and IV maintained their improvement in scores following induction therapy (Supplementary Figs 1, 2). In all patients, WPAI-UC instrument scores decreased or stayed the same by week 6 following open-label vedolizumab IV induction at weeks 0 and 2 (Supplementary Fig. 3). During maintenance treatment, WPAI-UC scores increased for patients on placebo, while patients on vedolizumab SC and IV maintained the improvement in scores they had achieved following induction treatment (Supplementary Fig. 3).
4. Safety
In the induction phase, 2 patients discontinued treatment with vedolizumab IV 300 mg due to a pretreatment event or AE. In the maintenance phase, planned study drug injections were completed by 8 out of 10 patients (80%) in the vedolizumab SC group and 2 out of 10 patients (20%) in the placebo group; and planned infusions were completed by 1 out of 2 patients (50%) in the vedolizumab IV group. The primary reason(s) for discontinuation was lack of efficacy in the vedolizumab SC and placebo groups, and pretreatment event or AE in the vedolizumab IV group.
In the induction phase, treatment-emergent AEs (TEAEs) were reported in 20 out of 27 patients (74%) (Table 4). The most frequent TEAE was nasopharyngitis (Table 5). Drug-related TEAEs occurred in 1 out of 27 patients (4%) (Table 4). Most AEs were mild to moderate and no deaths occurred. Serious TEAEs occurred in 3 out of 27 patients (11%). TEAEs leading to discontinuation occurred in 2 out of 27 patients (7%). There were no injection-site reactions (Table 5).
In the maintenance phase, TEAEs were reported in 21 out of 22 patients (96%), as follows: vedolizumab SC, 9 out of 10 patients (90%); placebo, 10 out of 10 patients (100%); and vedolizumab IV, 2 out of 2 patients (100%) (Table 4). The most frequent TEAE was nasopharyngitis (Table 5). Drug-related TEAEs were as follows: vedolizumab SC, 3 out of 10 patients (30%); placebo, 4 out of 10 patients (40%); and vedolizumab IV, 1 out of 2 patients (50%) (Table 4). Most AEs were mild to moderate and no deaths occurred. Serious TEAEs were as follows: vedolizumab SC, 1 out of 10 patients (10%); placebo, 1 out of 10 patients (10%); and vedolizumab IV, 1 out of 2 patients (50%). TEAEs leading to discontinuation occurred in 1 out of 2 patients (50%) in the vedolizumab IV group and in no patients in the other 2 treatment groups. Two patients who received vedolizumab SC experienced an injection-site reaction (20%) compared with none in those who received placebo or vedolizumab IV (Table 5).
5. Immunogenicity
Following vedolizumab IV induction therapy, AVAs were detected in 10% (1/10) of patients who received vedolizumab SC, 30% (3/10) of patients who received placebo, and in no patients who received vedolizumab IV. None of the AVA-positive patients who received vedolizumab SC were persistently positive or developed neutralizing antibodies. Among AVA-positive patients on placebo, 67% (2/3) were persistently positive and 100% (3/3) developed neutralizing antibodies. No discernable relationship was observed between AVA status and safety issues that related to injection-site reactions or hypersensitivity reactions (Supplementary Table 3).
DISCUSSION
The results of this ad-hoc analysis are limited by the small sample size; nevertheless, the results indicate that the efficacy of vedolizumab SC compared with placebo in a subgroup of Japanese patients with UC is consistent with the overall VISIBLE 1 study population. The results for vedolizumab and placebo arms in the subgroup of Japanese patients and overall VISIBLE 1 study population were similar at week 52 for clinical remission, endoscopic improvement, durable clinical response and durable clinical remission, with the exception of corticosteroid-free remission.
The clinical response rate at week 6 (45%) with vedolizumab IV in the present study was similar to the global VISIBLE population at week 6 (56%) [17]. In addition, the higher clinical remission rate at week 52 in patients who were TNF-α antagonist therapy naïve and received vedolizumab SC compared with placebo (60% vs. 25%, respectively) is consistent with that reported at week 60 in Japanese patients who received vedolizumab IV compared with placebo (54% vs. 36%, respectively) [11].
The safety and tolerability of vedolizumab SC in patients with UC was generally favorable with no safety issues that limited treatment, and was consistent with the overall VISIBLE 1 population [17]. In Japan, the SC administration of other biologics in patients with UC or CD has also been reported as safe, with similar safety profiles to their overall study populations [1,19]. Injection-site reactions occurred in 2 patients (20%) who received vedolizumab SC and in no patients who received vedolizumab IV or placebo. In the present analysis, the rate of injection-site reactions observed in patients who received vedolizumab SC was broadly in line with that in the overall VISIBLE 1 study population [17] and with other SC treatments for patients with IBD (ranging from 3% to 20%) [20-22].
The proportion of patients who received vedolizumab SC and were positive for AVA was consistent with that reported for the overall VISIBLE 1 population [17] and the pooled GEMINI 1 and GEMINI 2 population [23]. The proportion of patients who received vedolizumab SC and were positive for AVA (10%) was within the range of anti-drug antibody formation rates (0–65%) observed with other biologics in patients with IBD [24]. No relationship was observed between AVA status and infusion-related reactions and hypersensitivity reactions.
PROs improved following vedolizumab IV during induction therapy. While improvements in PROs were maintained throughout the maintenance treatment phase with vedolizumab SC and IV, they were not maintained with placebo. The improvements in PROs were consistent with results for efficacy, safety, and tolerability endpoints, and may benefit patients’ perceived quality of life. The improvements in PROs in Japanese patients who received vedolizumab SC were also consistent with those in health-related quality of life measures in the overall VISIBLE 1 study population who received vedolizumab SC [17], those in patients with UC in GEMINI 1 who received vedolizumab IV [25], and those in Japanese patients with other biologics [26,27].
The results of the present analysis indicate that vedolizumab SC may provide a similar level of clinical benefit to that with vedolizumab IV in terms of long-term efficacy and safety, and indicate that both routes of administration are also viable options for Japanese patients. Recently, the phase 3b randomized, double-blind VARSITY study evaluated the efficacy and safety of vedolizumab IV compared with adalimumab SC at week 52 in patients with moderate to severe UC [28]. The primary endpoint was met with a significantly higher proportion of patients with clinical remission (complete Mayo score ≤ 2 with no subscore > 1) at week 52 in the vedolizumab group (31%) than the adalimumab group (23%; P=0.0061) [28]. Head-to-head studies of vedolizumab SC with other biologics administered via the SC route are required.
A nationwide web-based survey on preferences for shared decision making (SDM) was conducted in Japanese patients with IBD [29]. Results showed that patients receiving biologics significantly favored SDM compared with those not receiving biologics, that patients with history of surgery significantly favored SDM compared with those without, and that most patients treated at university hospitals favored SDM [29]. In addition, a clear correlation was observed between the level of agreement that patients and their doctors had regarding SDM and patients’ overall satisfaction with their treatment [30]. Having both SC and IV routes of administration as available treatment options may help physicians to tailor treatment decisions and thus maximize patient compliance and improve clinical outcomes. Further surveys on routes of administration, patient preferences, and outcomes are needed to inform physicians in Japan.
Studies both in and outside of Japan have evaluated patient satisfaction and patient adherence with regard to biologics. A multicenter questionnaire survey was conducted in Japanese patients with CD to assess their satisfaction with and adherence to self-injections of adalimumab [31]. Results showed that 38% of patients would not accept self-injection therapy before treatment initiation; however, 75% of patients were satisfied with the treatment after treatment initiation [31]. In addition, patient adherence to self-injection therapy with adalimumab was high at 85% [31]. A hospital claims database analysis in Japanese patients with rheumatoid arthritis indicated that while the administration route does not have a significant association with the initiation of biologics, it does play a major role in switches between biologic agents [32]. A post-marketing surveillance study of self-injection of methotrexate SC in German patients with rheumatoid arthritis or psoriatic arthritis found that self-injection led to patients feeling more independence (89%) and a better quality of life (84%) [33]. Finally, a retrospective, longitudinal analysis of a cohort of patients with UC in the Japan Medical Data Center claims database evaluated the long-term persistence associated with adalimumab or infliximab combined with an immunomodulator (administered as step-up treatment or simultaneously) compared with adalimumab or infliximab alone during maintenance therapy. Results showed that in patients with UC who had been newly prescribed either infliximab IV or adalimumab SC, the proportion who switched treatment was similar at ~10% [34]. However, this study did not investigate the motivational aspects of the switch [34]. These studies, including those from other disease areas, suggest that SC administration could be a useful option to meet various individual preferences and potentially improve patient satisfaction.
A limitation of this post hoc analysis is the small number of Japanese patients enrolled in the VISIBLE 1 study, which prevented meaningful analyses of pharmacokinetics and of the vedolizumab IV subgroup. This limitation was compounded by the overall sample size of VISIBLE 1, which was smaller than GEMINI 1 study of vedolizumab IV in patients with UC [13]. In addition, this limitation may have contributed to the lack of statistically significant differences (despite numerically greater differences) between treatment groups for some efficacy endpoints, as well as the numerically lower proportion of patients with corticosteroid-free clinical remission in the vedolizumab SC group than in the placebo group. Analysis in a larger number of Japanese patients would also enable the efficacy of vedolizumab in those with prior TNF-α antagonist use to be determined. Another limitation is that outcomes of SC maintenance were examined in week 6 responders, after only 2 induction infusions. Therefore, this overall study may have selected patients who responded very well to vedolizumab, and in turn influenced the results of the present analysis. Further study is also likely to be needed to determine the rate of injection-site reactions with vedolizumab SC.
In conclusion, the new vedolizumab SC formulation (108 mg Q2W) exhibited similar trends in efficacy as maintenance therapy in Japanese patients with UC compared with the overall VISIBLE 1 study population, as did vedolizumab IV as induction therapy [17]. The safety and tolerability of vedolizumab SC were generally similar to the whole study population [17]. Further study of vedolizumab SC in a prospective, specifically designed study of a larger number of Japanese patients with UC is warranted, as well as in patients with UC who are not Japanese or Caucasian.
 XML Download
XML Download