

 PDF
PDF Citation
Citation Print
Print



INTRODUCTION
Vedolizumab is a humanized immunoglobulin G1 (IgG1) monoclonal antibody that selectively targets human lymphocyte α4β7 integrin on the surface of a subset of leukocytes, which mediates lymphocyte trafficking to gastrointestinal mucosa and gut-associated lymphoid tissue through mucosal addressin cell adhesion molecule-1 (MAdCAM 1) [1]. The novel mechanism of action of vedolizumab allows it to bind exclusively to the α4β7 integrin, antagonizing its adherence to MAdCAM-1 and thereby impairing the migration of leukocytes into gastrointestinal mucosa [1]. The gut-selective, anti-inflammatory activity of vedolizumab enables targeted therapy without generalized immunosuppression. Vedolizumab intravenous (IV), a lyophilized formulation of vedolizumab intended for IV infusion has been granted marketing approval in over 65 countries worldwide, including the United States, European Union, and Japan. Vedolizumab IV is indicated as a first-line biologic for adult patients with moderately to severely active ulcerative colitis (UC) or Crohn’s disease (CD) who have failed to respond to or are intolerant of conventional therapy; or as a secondline biologic following treatment with a tumor necrosis factor (TNF) antagonist [2,3]. Vedolizumab IV is well-established as effective and safe for both induction and maintenance treatment of UC and CD.
The recommended dose regimen for vedolizumab IV is 300 mg administered by IV infusion at 0, 2, and 6 weeks and then every 8 weeks (Q8W) thereafter. In some regions, increasing the frequency of dosing to every 4 weeks (Q4W) is indicated for those who have lost response to the Q8W regimen during the maintenance phase. Therapy for UC or CD patients should be discontinued if no evidence of therapeutic benefit is observed by week 14.
The pharmacokinetics (PK) of vedolizumab and other therapeutic monoclonal antibodies used for the treatment of UC and CD have been previously reported [4-7]. Several factors are associated with accelerated clearance of these antibodies, including the presence of anti-drug antibodies, sex, body size, concomitant immunosuppressant use, inflammatory bowel disease (IBD) type, albumin concentration, and degree of systemic inflammation [8-10]. For vedolizumab, only extreme albumin and body weight values were identified in a previously reported population PK analysis as potential clinically important predictors of its linear clearance (CLL) in a predominantly Western population [4].
Biologic drugs have shown that they are generally insensitive to ethnicity; most monoclonal antibodies show no differences between ethnic groups with respect to PK [8,11-16]. For example, golimumab showed comparable PK in healthy Caucasian and Japanese subjects following a single dose subcutaneous administration [13]. Similar results were reported for certolizumab in patients with CD [16].
Here we report the results of a new analysis which aimed to update the previously developed population PK model for vedolizumab [4] with data from phase 3 studies in Japanese patients.
Because genetic, environmental, and/or cultural differences amongst populations may result in different PK and unexpected clinical response rates, the goal of this analysis is to assess the impact of covariates on vedolizumab PK in Asian and non-Asian UC and CD patients, while describing the typical population parameters as well as interindividual and residual variability.
METHODS
1. Study Design and Sample Collection
A similar approach to what was presented before was adapted in the current analysis, where the majority of the data consisted of sparse vedolizumab PK sampling from phase 3 studies [4]. The analysis was conducted using vedolizumab serum concentrations obtained from 5 randomized, placebo-controlled clinical studies: a phase 1 study in Japanese patients with active UC (CPH 001), two phase 3 studies in patients with moderately to severely active UC (GEMINI 1; NCT00783718, and CCT 101; NCT02039505), and two phase 3 studies in patients with moderately to severely active CD (GEMINI 2; NCT00783692 and CCT-001; NCT02038920) (Supplementary Table 1) [17-20]. Studies CPH-001, CCT-001 and CCT-101 were all conducted in Asian (Japanese) patients, whereas the GEMINI studies were conducted in predominantly Western populations. The study designs and clinical data for the CPH-001, GEMINI 1 and 2 studies have been previously reported [18-20]. The study designs for CCT-001 and CCT 101 were consistent with those of the GEMINI studies, but were conducted in Japanese patients [21]. All study protocols and consent forms were approved by institutional review boards or ethics committees at the study sites, and studies were conducted in accordance with the principles of good clinical practice and the Declaration of Helsinki. All patients provided written informed consent before study participation.
Details of the blood sampling times for PK and anti-vedolizumab antibodies (AVA) are provided in Supplementary Table 1. Extensive PK sampling was used in the phase 1 study, whereas sparse sampling was used in the phase 3 studies.
2. Assays
Vedolizumab serum concentrations were determined using a sandwich enzyme-linked immunosorbent assay (ELISA) as described previously [4]. The assay used to quantify vedolizumab serum concentrations in studies GEMINI 1 and 2 and CPH 001 had lower and upper limits of 0.125 µg/mL and 8 µg/mL, respectively. The assay used in studies CCT-001 and CCT-101 had a calibration range of 0.200 µg/mL to 8 µg/mL.
The presence of AVA in human serum samples was detected using a validated electrochemiluminescence (ECL) assay. All samples that screened positive were further diluted to determine the final AVA titer using standard techniques. If both screening dilutions were negative, the sample was considered negative.
3. Data Assembly
The dosing, covariate, and PK data were merged and formatted for the population analysis using R, version 3.3.3 of R (R Foundation for Statistical Computing, Vienna, Austria; http://www.r-project.org/). Concentration measurements with missing observation times had times imputed relative to dosing times. Pre-first dose observations with measurable concentrations were excluded from the analysis but maintained in the dataset. Patients who received placebo in both induction and maintenance phases from GEMINI 1, GEMINI 2, CCT-101, and CCT-001 were not included in the dataset. Samples with vedolizumab concentrations below the limit of quantification (BLQ) were not evaluated during the population PK model development given that the percentage of BLQ data after the first dose was small (6% of the total data), although these sample records were maintained in the dataset for model evaluation purposes.
Covariates present in the population PK dataset were body weight, age, sex, race, IBD diagnosis, serum albumin, and AVA titer. IBD diagnosis was defined as UC or CD. Six different race categories were present in the dataset: white, Asian, American Indian or Alaska Native, Native Hawaiian or other Pacific Islander, and other. Body weight, albumin and AVA titer were present in the dataset as time-varying covariates. All other covariate data present in the dataset were baseline values. The AVA-related variables in the analysis dataset represented total binding antibodies (not neutralizing antibodies).
Body weight and serum albumin were typically observed on a frequent basis over the course of the studies and therefore, interpolation of values between observed time points was done using a last observation carried forward approach rather than, for example, linear interpolation. Body weight and albumin were often observed on dosing or PK sampling visits. AVA titers were sampled much less frequently compared to body weight and albumin, and so AVA titers were imputed between observed values using linear interpolation. This approach was taken to avoid unreliable and biased covariate estimates due to NONMEM backfilling covariate records between two time points during the estimation procedure.
An occasion variable named OCC was included in the dataset for modeling interoccasion variability (IOV). An occasion was defined as a dosing interval with at least one associated vedolizumab PK measurement.
4. Population Pharmacokinetic Model Development
The population PK analysis for repeated measures was conducted using a nonlinear mixed-effects modeling approach (NONMEM 7, version 7.3; ICON Development Solutions, Ellicott City, MD, USA) [22]. The pre- and post-processing input/out and analysis scripting was programmed using R. The Monte Carlo importance sampling expectation-maximization (EM) method was employed during model development, and the final population PK model was fit to the data using the full Bayesian Markov Chain Monte Carlo (MCMC) method. The Bayesian prior distributions specified in the final population PK model were utilized during all stages of model development. Both Bayesian MCMC and importance sampling methods were implemented with the $PRIOR subroutine to incorporate priors into the population PK models [4].
In the previous vedolizumab population analysis, the base population PK model was developed using extensively sampled PK data from studies C13002 and C13009. Results from the base population PK model were then used as prior information to selectively inform a subset of PK parameters in the final population model fit to sparse phase 3 data (GEMINI 1, 2, and 3) [4]. A similar approach was adapted here, where the majority of the data for the final population model consisted of sparse vedolizumab PK sampling from phase 3 studies GEMINI 1 and 2, CCT 101 and CCT-001.
The final population PK model developed previously, was used as a starting point for model development in the current analysis [4]. Of note, the previous published population model was developed with AVA as determined by an ELISA assay [4]. The samples were subsequently re assayed with an ECL assay and the population PK model was updated. Because of interference issues with the ELISA assay, the impact of AVA on vedolizumab CLL in the previous analysis was investigated using a subject-level AVA variable (subject never had AVA-positive result versus subject had at least one AVA-positive result). The new analysis had updated ECL assay data and time-varying AVA data in the dataset, so other functional forms of modeling the AVA effect on CLL were evaluated using the continuous titer data (rather than a categorical effect as previously) [23].
Prior distributions specified in the previous final population PK model were incorporated into the current analysis model development. In brief, informative priors were defined for maximum elimination rate (Vmax), concentration at half-maximum elimination rate (Km), peripheral volume distribution (Vp) and intercompartmental clearance (Q), whereas uninformative (vague) priors were defined for the remaining fixed-effect parameters (structural PK parameters and covariate coefficients) and interindividual random-effect parameters in the model. This was implemented in NONMEM using the $PRIOR subroutine. The uninformative priors allowed estimation of these parameters to be driven by study data in the current analysis rather than previous results.
Covariate effects in the previous final model were removed and the full covariate model was reconstructed to incorporate the covariates of interest for this analysis. Covariates were removed from the previous model for several reasons: not of clinical interest for the current analysis, was found not to be a clinically meaningful covariate in the prior analysis, or was not measured in the Japanese studies (CPH-001, CCT-101, and CCT 001).
Body weight was chosen to represent changes in vedolizumab PK as a function of body size and was described using an allometric model with a reference weight of 70 kg. The other continuous covariates entered the model as power functions normalized by a reference value. The categorical covariates entered the model as power functions, with a separate dichotomous (0, 1) covariate serving as an on-off switch for each effect.
Inferences about the clinical relevance of covariates were based on the resulting parameter estimates and measures of estimation precision (Bayesian 95% credible intervals [CDIs]) from the full model. In the absence of an exposure-response relationship for efficacy-related clinical endpoints to provide a context for interpretation of PK variability, covariate effect sizes on CLL greater than ± 25% of the normalized reference value were proposed as clinically meaningful changes.
Further details regarding the population PK analysis methods, including modeling assumptions and model evaluation, were provided in the supplementary appendix of Rosario et al. (2015) [4]. Further details regarding the general modeling methods for this analysis are provided in Supplementary Material 1.
5. Population PK Model Evaluation
The final population PK model and parameter estimates were investigated with a posterior predictive check method. The basic premise is that a model and parameters derived from an observed dataset should produce simulated data that are similar to the original observed data. Five hundred random samples of parameter estimates from the joint posterior probability distribution were taken. For each set of parameter estimates, one simulation replicate of the original dataset was generated using the final population PK model. Distributions of an exposure metric of the simulated data were compared with the distribution of the same exposure metric in the observed dataset using a visual predictive check (VpC) for study CPH-001 or quantile plots for the remaining studies. A VpC was chosen for CPH-001 as this study had more intensive PK sampling compared to other studies in the analysis which had sparse sampling.
The precision of the parameter estimates from the final population PK model was assessed by summarizing the parameter samples from the Bayesian posterior probability distributions. Bayesian 95% CDIs were constructed by observing the 2.5th and 97.5th percentiles of the posterior distributions. No sample thinning was performed when summarizing the posterior distributions.
RESULTS
1. Pharmacokinetic Analysis Dataset
The PK analysis dataset consisted of 1,933 individuals who contributed 16,598 evaluable vedolizumab serum samples, including 9 patients from the phase 1 study (CPH-001, UC), and 743, 966, 152, and 63 patients from the phase 3 GEMINI 1 (UC), GEMINI 2 (CD), CCT-101 (UC), and CCT-001 (CD) studies, respectively.
Demographics and other characteristics of the PK analysis dataset are summarized in Table 1. The dataset consisted of 1,018 men (53%) and 915 women (47%) with ages ranging from 17 to 79 years and baseline body weights ranging from 28 to 172 kg. Serum baseline albumin ranged from 1.40 to 5.30 g/dL. A total of 1,029 individuals had CD (53%) and 904 (47%) had UC. The dataset consisted of 77% white patients, and 396 Asian patients (20%).
There were 1,774 patients (92%) with observed AVA titer that were all negative and 159 patients (8.2%) with at least one observed positive AVA titer. Titer values for positive samples were typically between 10 and 6,250 with very few titers above 6,250.
The effect of race on vedolizumab CLL was modeled using 2 categories: Asian and non-Asian. American Indian, black, Native Hawaiian and other patients were pooled together with white patients to define the non-Asian category given the small number of patients in those racial groups. Japanese patients in studies CPH-001, CCT-101, and CCT-001 (n = 224) made up 57% of the total number of Asian patients (n = 396).
2. Population Pharmacokinetic Modeling Results
1) Final PK Model
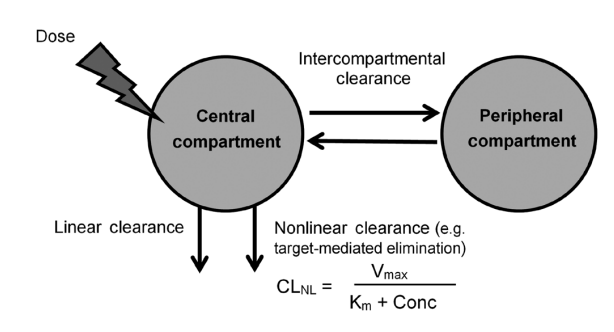
The structural component of the final vedolizumab population PK model consisted of a two-compartment model with zero order (IV infusion) input and parallel linear and nonlinear elimination pathways. The model was parameterized in terms of CLL, central volume distribution (Vc), Vp, Q, Vmax, and Km (Fig. 1). The model was implemented using PREDPP subroutine ADVAN13 and TRANS1. Interindividual random effect distributions were modeled using exponential variance models for CLL, Vc, and Vp, with a full block covariance matrix, while residual random effects were described with a proportional error model. IOV was modeled on CLL using an exponential random effect.
The primary covariates of interest for the full covariate model were defined a priori. They included: body weight as a predictor of CLL, Vc, Vp, Q, and Vmax; serum albumin as predictor of CLL; AVA as a predictor of CLL; race (Asian vs. non-Asian) as predictor of CLL; and diagnosis (UC vs. CD) as a predictor of CLL. All covariates (with exception of race and diagnosis) were time-varying.
CLL, Vc, Vp, Q, and Vmax were scaled by body weight using a power model normalized to a weight of 70 kg. The power coefficients were estimated for CLL and Vc and fixed to theoretical allometric values for the remaining parameters (0.75 for Q and Vmax; 1 for Vp). The effect of albumin on CLL was described using a power model normalized to albumin concentration of 4.0 g/dL. The effect of race (Asian vs. non-Asian) on CLL was described using a multiplicative function with the covariate serving as an indicator variable for the effect. The effect of diagnosis (UC vs. CD) on CLL was similarly described by a multiplicative function with the covariate serving as an indicator variable. The effect of AVA on CLL was investigated by modeling separate typical value parameters of CLL for AVA-negative and AVA-positive patients. The change in CLL as a function of AVA titer was described using a log-transformed power model normalized to a titer of 250. Negative AVA was defined as a titer < 10 and positive AVA was defined as a titer ≥ 10. The separate typical CLL values for AVA-negative and AVA-positive shared the same interindividual variance term and covariate effects (i.e., body weight, albumin, race and diagnosis).
Diagnostic plots demonstrated that the final model was consistent with the observed data and showed an adequate fit of the model (Supplementary Figs 1, 2). When stratifying plots of observed data vs model predictions by race (Supplementary Fig. 2), diagnosis or AVA titer (not shown), no notable trends of systematic bias were observed. This indicated that the model adequately described the data across different races, diagnoses and AVA titers.
PK parameter estimates and 95% CDIs from the final population PK model and their interindividual variability estimates are shown in Table 2. Typical value estimates of the PK parameters are presented for the reference covariates (body weight: 70 kg, albumin: 4 g/dL, race: non-Asian, diagnosis: UC, AVAnegative titer: < 10; AVA-positive titer: 250). The point estimates in the table were derived by observing the median of the Bayesian posterior distributions. The linear elimination phase half life (t1/2) was 24.7 days for AVA-negative patients and 18.1 days for AVA-positive patients (titer 250). Interindividual variability estimates were indicative of moderate to large unexplained variability. The interindividual variability estimates (percent coefficient of variation [%CV]) from the model were 30.8% for CLL, and 20.2% for Vc, and 70.2% for Vp (Table 2). IOV on CLL was moderate, with an estimated %CV of 20.3%. The residual error (unexplained random residual variability in the model) was relatively small (%CV of 17.8%) (Table 2).
2) Final Model Covariate Effects
The effect of body weight was a determinant of variability in CLL with an estimated power coefficient (95% CDI) of 0.472 (0.400, 0.533) (Table 2). Serum albumin was also a determinant of variability in CLL with an estimated power coefficient (95% CDI) of –1.19 (–1.27, –1.11).
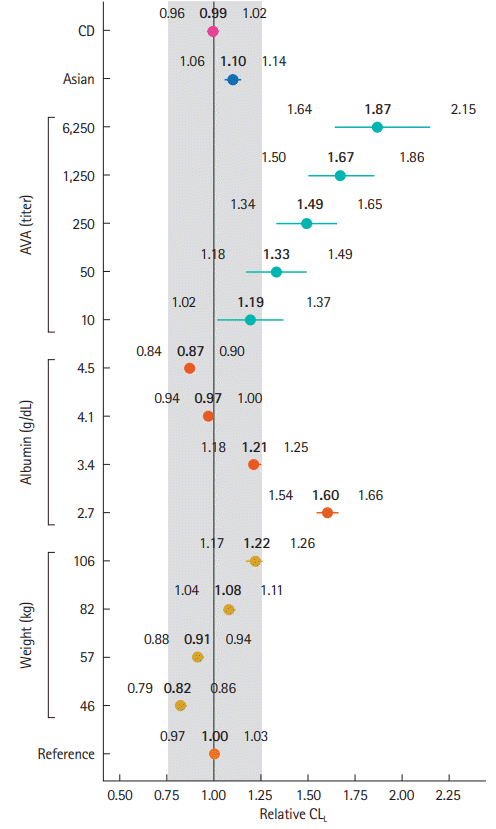
The effect of race on CLL was small and precisely estimated with a point estimate (95% CDI) indicating an increase in CLL in Asian patients by a factor of 1.10 (1.06, 1.14) compared to non-Asian patients (Table 2).
There was no effect of IBD diagnosis on CLL; the effect was precisely estimated with a point estimate (95% CDI) indicating a decrease in CLL in CD patients by a factor of 0.990 (0.960, 1.020) compared to UC patients.
The presence of AVA was estimated to increase vedolizumab CLL. For a typical AVA positive patient with a titer of 250, the point estimate and 95% CDI for CLL was 0.246 L/day (0.222, 0.273) compared to 0.165 L/day (0.160, 0.170) for the reference patient with a negative AVA. The 95% CDI for the power coefficient of the AVA titer effect was relatively wide (0.0404, 0.1020). This imprecision is in part likely due to the low incidence of positive AVA titers in the dataset.
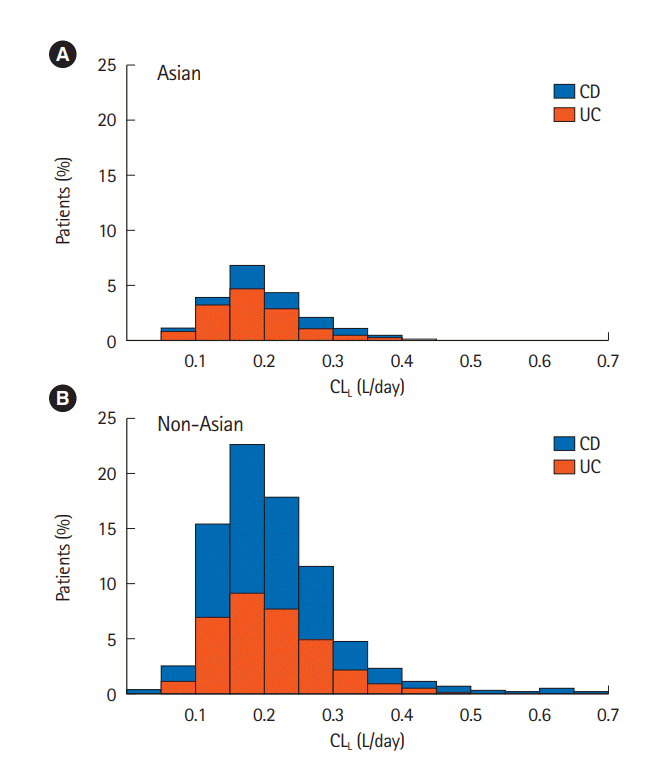
Further evaluation of covariates on CLL was conducted via simulation as described previously [4]. The impact of covariates on vedolizumab CLL is shown in Fig. 2. Covariate effect sizes of ± 25% from the typical reference patient were used as a limit for clinically meaningful changes. The figure clearly shows the expected impact of body weight, albumin and AVA on CLL. Body weight and albumin appeared not to have a clinically meaningful impact across the range of values evaluated except at the extremes (i.e., observed 5th and 95th percentiles of the covariates). Extreme values of body weight (e.g., 106 kg) and albumin (e.g., 2.7 g/dL) and a positive AVA titer ( ≥ 10) were identified as potentially clinically important predictors of CLL, as the 95% CDI for the covariate effect partially or completely fell outside of the ± 25% range. The effect of race was not considered to be clinically important as the entire 95% CDI for the covariate effect fell within the ± 25% range. Similarly, there was no clinically meaningful effect of IBD diagnosis on CLL. The distribution of individual patient vedolizumab CLL estimates from the final population PK model for Asian and non-Asian in patients with UC and CD are shown in Fig. 3. This figure reflects the minimal impact of race and IBD diagnosis on vedolizumab CLL.
3. Pharmacokinetic Model Evaluation
The vedolizumab population PK model evaluation results, which included the results of a posterior predictive check and Bayesian 95% CDIs, revealed that the final model provided a reliable description of the data with good precision for most fixed and random effect parameters.
The precision of the population PK model parameters was assessed by summarizing the parameter samples from the Bayesian posterior probability distributions. Bayesian 95% CDIs were constructed by observing the 2.5th and 97.5th percentiles of the posterior parameter distributions. Overall, most structural model parameters, covariate effects, and random effect variance terms were estimated with good precision (Table 2).
The final model and parameter estimates were evaluated with a simulation-based posterior predictive check method. Exploratory graphs (VpC and quantile-quantile plots) were generated to compare distributions of an exposure metric in the simulated data with the distribution of the same metric in the observed dataset (Supplementary Figs 3, 4; studies CPH-001, CCT-001, and CCT-101). The results demonstrated good agreement between observed and simulated data when evaluated under different vedolizumab regimens, race groups and IBD diagnosis. The predictive check results are not shown for the GEMINI studies but were consistent with those previously reported [4].
DISCUSSION
A population PK model for vedolizumab administered IV to patients with moderately to severely active UC and CD was developed, using data collected in clinical trials in Japan and outside Japan. The PK of vedolizumab was well described by a two-compartment model with zero-order input and parallel linear (CLL) and nonlinear (CLNL) elimination from the central compartment. The physiological interpretation of this model has been provided elsewhere [4]. The estimated half-life in AVAnegative patients was 24.7 days, which is consistent with that of endogenous human IgG1 (~21 days) and of therapeutic monoclonal antibodies of the IgG1 subclass [8]. The previously reported half-life of vedolizumab in non-Japanese patients was 25 days [4].
Previously we reported the effects of intrinsic and extrinsic covariates (body weight, age, albumin, fecal calprotectin, Crohn’s Disease Activity Index score, partial Mayo score, concomitant immunosuppressive therapy use, AVA status, and prior TNF-α antagonist therapy status) on vedolizumab CLL. In the analysis reported here, we only carried forward and evaluated the effects of body weight and albumin, which were identified as potentially clinically important covariates in the previous analysis. AVA titer, race, and IBD diagnosis were also of clinical interest for this analysis and thus included in the full covariate model.
For the first time, we assessed the effect of race (Asian vs. non-Asian) on the elimination of vedolizumab. Race was not explored in the previous analysis. The impact of race on clearance of vedolizumab was found to not likely be clinically significant. The effect of race on CLL was small and precisely estimated with a point estimate (95% CDI) indicating an increase in CLL in Asians by a factor of 1.10 (1.06, 1.14) compared to non-Asian patients. The effect of Asian race on CLL was estimated with good precision and the effect 95% interval lied within the ± 25% region of unimportance. If there was insufficient information on this effect (e.g., sample size) we would expect the 95% interval of the posterior distribution for the covariate effect to be wide and span across values of a covariate effect that are both clinically important and unimportant.
Likewise, a comparison of the distribution of individual patient CLL estimates between Asian and non-Asian patients showed similarities between the 2 race groups (Fig. 3). These results are expected as vedolizumab is an antibody, and IgG1 characteristics are generally insensitive to ethnicity. Beyond the PK data presented here, phase 3 trials in Asian UC or CD patients treated by vedolizumab at a similar posology have shown similar trends in clinical efficacy and safety to the GEMINI studies [18,19,21].
Extreme values of albumin had important effects on vedolizumab CLL. Albumin concentrations below 2.7 g/dL were associated with increased vedolizumab CLL relative to a patient with 4 g/dL albumin that was greater than the prespecified criterion for clinical significance (assuming reference values for all other covariates). Similar association of low albumin concentrations with increased clearance has been reported in population PK analyses of other monoclonal antibodies [9,10]. A potential physiological mechanism behind the relationship between albumin and vedolizumab CLL has been discussed elsewhere [4].
The second important covariate identified was body weight, which was positively correlated with vedolizumab CLL, but not considered clinically relevant when evaluated over a range of weights reflecting the majority of observed weights in the dataset. For example, a patient of 46 kg with reference values for all other covariates had an estimated (95% CDI) relative change in CLL of 0.82 (0.79, 0.86) versus a 70 kg patient; this result is within the prespecified criterion for clinical significance (Fig. 2). Likewise, a patient of 106 kg had an estimated (95% CDI) relative change in CLL of 1.22 (1.17, 1.26) versus a 70 kg patient; a result that just crossed the criterion for clinical significance. The impact of body weight on vedolizumab CLL is consistent with that reported in population PK analyses of other therapeutic monoclonal antibodies [8,10].
Previously, we reported the impact of AVA on clearance of vedolizumab using data from an ELISA assay [4]. Later, samples from GEMINI 1 and 2 were re-assayed using an ECL assay and the population PK model was updated [23]. In this updated model the covariate model for AVA on CLL was also revised. Here we reported the effect of AVA on vedolizumab CLL using the new assay and the updated model. In the current analysis, the presence of AVAs increased CLL from 0.165 to 0.246 L/day, for AVA-negative patients ( < 10 titers) and AVA-positive patients (titer 250), respectively. The estimated half-life was 24.7 days for the linear elimination phase for AVA-negative patients ( < 10 titers) and 18.1 days for AVA-positive patients (titer 250). The impact of AVA on CLL reported here was greater than that previously reported [4]. This can be attributed to a number of factors outlined above and in section “Population pharmacokinetic model development.”
There was no effect of IBD diagnosis on CLL; the point estimate (95% CDI) indicated a lower CLL in CD patients by a factor of 0.990 (0.960, 1.020) compared to UC patients. Further evaluation of the effect of IBD diagnosis on CLL showed that this covariate is not expected to be clinically meaningful (Fig. 2). This finding is consistent with the previous population PK analysis of vedolizumab [4].
In conclusion, a population model characterizing the PK properties of vedolizumab was successfully developed for Asian and non-Asian patients with UC or CD. The modeling results suggest that vedolizumab CLL is similar in Asian patients compared to non-Asian patients and in UC and CD patients. Albumin, body weight and AVA were identified as predictors of vedolizumab CLL, but the effects of these covariates are only considered clinically meaningful at extreme values. This analysis supports the use of flat-fixed dosing with vedolizumab in Asian and non-Asian UC and CD patients.
 XML Download
XML Download