

 PDF
PDF Citation
Citation Print
Print



INTRODUCTION
The large intestine is built with a single layer of epithelial cells showing inward invaginations or folds called crypts. Crypts are lined with absorptive and mucus producing goblet cells, entero-endocrine cells and only in cecum and proximal right colon, with Paneth cells [1]. The crypts replicate by symmetric fission, beginning at their base, and proceeding upwards until 2 identical, individual crypts are created [2,3]. In well-oriented sections, a characteristic crypt assemblage of tightly packed crypts, parallel as “rows of test tubes” perpendicular to the surface-epithelium-muscularis cells mucosae-axis, are found [4]. A slight variation in the configuration of the crypts and in the space between the crypts may occur, but crypt branching is rarely seen in fixed preparations [4-6].
In a previous study, we found only 3.7 (range, 2–5) single colon crypts lined with normal epithelium but exhibiting asymmetric shapes; called colon crypts with corrupted shapes (CCS), they were found amongst more than 200,000 crypts investigated in 22 colon segments, proximal or distal to surgically removed colon cancer in humans [7]. The remnant crypts in the colon segments in the 22 patients showed crypts with normal shapes. These findings indicated that the vast majority of the normal mucosa of patients having synchronously a colon cancer, displays crypts with normal shapes and that CCS very rarely occur [7], thus resembling the normal mucosa in individuals without colon cancer [1,5,6].
RISK FACTORS IN THE DEVELOPMENT OF COLON NEOPLASIAS
The vast mucosal area of the human large bowel (estimated at 2 m2) [8] is relentlessly exposed to a series of risk factors able to generate neoplastic lesions. The main risk factors for the development of neoplastic lesions in the colonic mucosa are: (1) sex (the risk is higher in males), (2) genetic differences (the risk is higher in patients with germline mutations in one of the DNA mismatch repair genes), (3) obesity (physical inactivity and obesity are strong independent determinants associated to colon cancer), (4) type 2 diabetes (hyperinsulinemia is important in the pathogenesis of colon cancer), (5) environmental carcinogens (such as polycyclic aromatic hydrocarbon when meat is heated above 180°C for long periods), (6) mutant gut microbiome, (7) breakdown of the gut macrophagebarrier (allowing the trespassing of gut microbiome into the host, destabilizing thereby host immunity in inflammatory bowel disease), and (8) life style factors (high use of alcohol and tobacco, known to encourage the development of sessile serrated adenoma/polyps) [9-15].
SPORADIC COLON ADENOMAS AND COLON CARCINOGENESIS
Under the influence of one or more of the aforementioned risk factors, the mucosa of the large intestine may undergo hubs of histological, biochemical and molecular aberrations, called adenomas [16-19]. Based on intraepithelial cellular alterations, adenomas are histologically divided into those with lowgrade dysplasia and with high-grade dysplasia [20]. Only those colonic adenomas breaking through the muscularis mucosa are considered as invasive carcinomas. Intramucosal invasion is not to be regarded as invasive carcinoma according to the World Health Organization (WHO) [20] and the Vienna Classification [21].
Triggered by the accumulation of molecular anomalies and epigenetic modification of gene expression [22-24], adenomas develop a particular propensity to progress to colon cancer. For many years, the general understanding was that the vast majority of colon cancer evolved from sporadic polypoid conventional (tubular or villous) adenomas-via the polypoid conventional adenoma-carcinoma pathway [25]. Subsequently, serrated adenomas [26] and sessile serrated adenoma/polyps [27] were incorporated as alternative pathways of colorectal carcinogenesis. It has been estimated that about 30% of all colorectal cancers (CRCs) progress via the serrated pathway [28,29]. In a study of all polypoid conventional adenomas registered at this hospital between 1993 and 2000 (n=3,135) [30] it was found that 93% were polypoid conventional adenomas, and 7%, serrated adenomas and microtubular adenomas [30,31]. Thus, at this hospital, polypoid conventional adenomas were also found to be the most commonly reported histologic phenotype of sporadic protruding adenomas.
In 1985 Muto et al. [32] detected at colonoscopy, small nonpolypoid mucosal lesions measuring up to 1 cm in diameter. At histological examination those non-polypoid mucosal lesions were found to be adenomas. Flat adenomas (FA), as they were called, were subsequently found associated with a more aggressive clinical behavior than their polypoid counterparts [33]. In 1997, Kudo et al. [34] reported non-polypoid (flat) colon neoplasms with a lateral diameter greater than 10 mm; they were called laterally-spreading tumors (LST). The biological significance of non-polypoid adenomas (i.e., FA and LST) was subsequently confirmed by an international group of Western and Japanese workers [35].
CRYPT WITH IRREGULAR SHAPES ASSOCIATED WITH COLONIC NEOPLASIAS
1. Aberrant Crypt Foci
There is much discussion concerning the significance of aberrant crypt foci (ACF) as precursors of colon cancer. ACF were first described by Bird [36] in 1987. Following staining of the surface of the colonic mucosa with methylene blue in carcinogentreated rodents, Bird found gross changes in the pit pattern consisting in groups of 5 or more colonic crypts.
In humans, Nascimbeni et al. [37] found that ACF occurred more frequently in patients with CRC than in controls, and claimed that the phenomenon sustained their putative role as a preneoplastic marker. Using methylene-blue-spraying and magnifying colonoscopy Drew et al. [38] identified in the human colon, ACF in the proximal colons of approximately 40% of individuals undergoing chromoendoscopy, concluding that ACF were the earlier morphologically identifiable lesion. In contrast, Fenoglio-Preiser and Noffsinger [39] wrote: “In humans, the frequency of ACF substantially outweighs the frequency of adenomas and carcinomas in the same colons, which suggests that most ACF may be innocent bystander lesions.” Dolara et al. [40] submitted that ACF and colon cancer risk seemed connected in Japan, but not in Europe or North America, and Ochiai et al. [41] claimed that the specificity of ACF as tumor precursors should be questioned, partly because of their predominant location in the proximal colon where tumors do not develop, and by their gradual disappearance over time. In addition, ACF frequently harbored K-Ras mutations, but rarely adenomatous polyposis coli mutations, strongly suggesting that the majority of ACF might not be true preneoplastic lesions [41]. More recently, Quintanilla et al. [42] concluded that rectal ACF in humans are not surrogate markers for CRC risk. Thus, 32 years after the initial report by Bird [36] in rodents, the significance of ACF as precursors of colon cancer in humans, remains controversial.
2. Crypts with Asymmetric Shapes Beneath Colon Adenomas
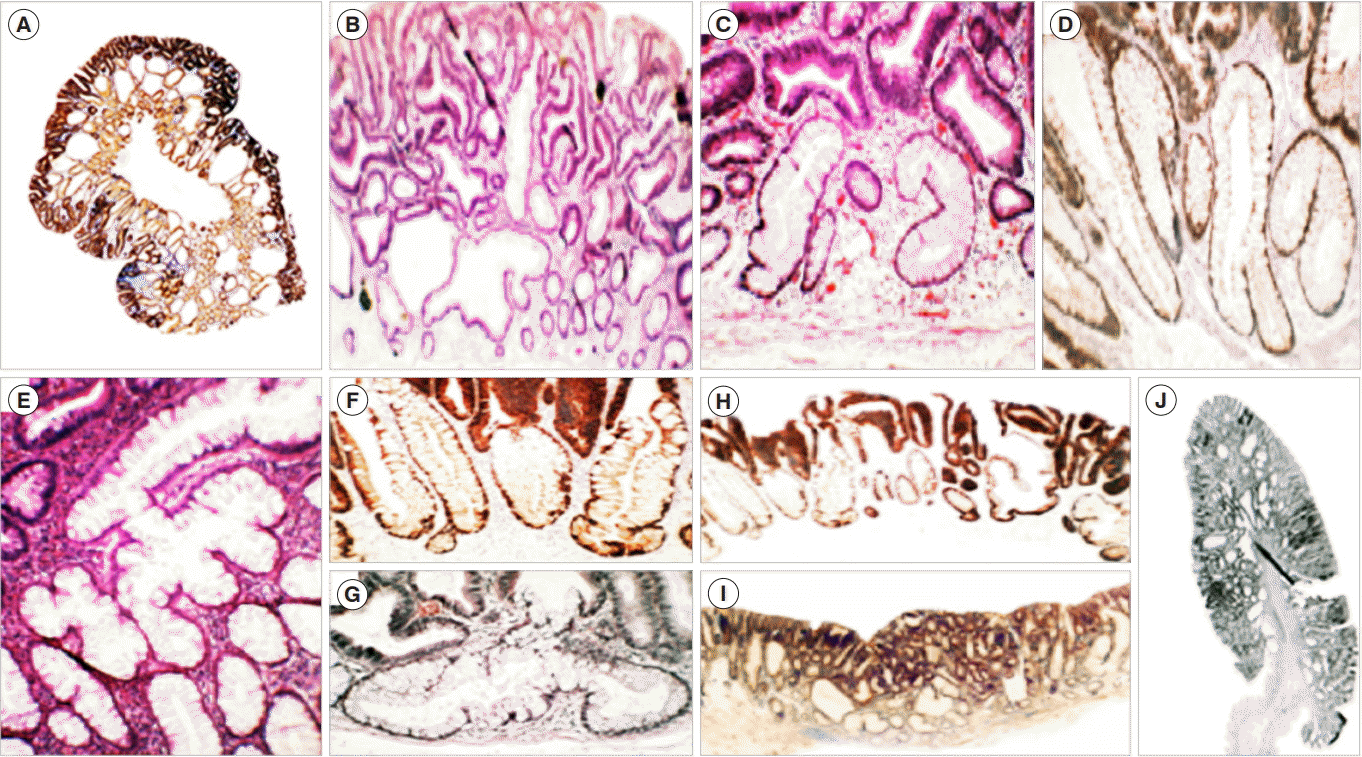
In contradistinction to ACF which are detected by observing the methylene blue-stained mucosa from the top, crypts with architectural distortions, also called crypts with CCS, are identified sidewise in well-oriented histological sections [7]. CCS in cluster were found assembled beneath the neoplastic canopy of 255 sporadic polypoid conventional adenomas [43]. CCS were characterized by fission distortions (exhibiting 2 or multiple non-identical crypt fissions), length distortions (hyperplasic crypts being more than twice the length of a normal crypt), outline distortions (crypts with multi-lobate outlines or cystic dilatations), and/or axial-polarity distortions in relationship to the muscularis mucosae (serpentine pattern, crypts, intercommunicating horizontal crypts, inverted L-shaped or ring-shaped horizontal crypts in “Indian files”) (Fig. 1). All sporadic polypoid conventional adenomas examined in that survey revealed CCS in cluster, assembled beneath the adenomatous tissue. Nearly three-quarters of the 255 sporadic, polypoid conventional adenomas had 3 or more CCS below and 46.3% revealed ≥4 CCS [43].
In another study, 51 colonic non-polypoid tubular adenomas were investigated [44]. Although the mean number of CCS was significantly higher in lateral spreading adenomas (≥10 mm) than in small, (“flat”) adenomas (≤9 mm), the mean CCS per mm ratio was 0.94 (range, 0.50–2.00) in FA and 0.92 (range, 0.40–2.93) in lateral spreading adenomas. Hence, the lateral expansion of non-polypoid colonic tubular adenomas seems to have evolved as a result of increased numbers of CCS, without increasing the number of CCS per mm [44].
3. Non-dysplastic Crypts with CCS Showing Redistributed Proliferating Cells and p53Upregulated Cells
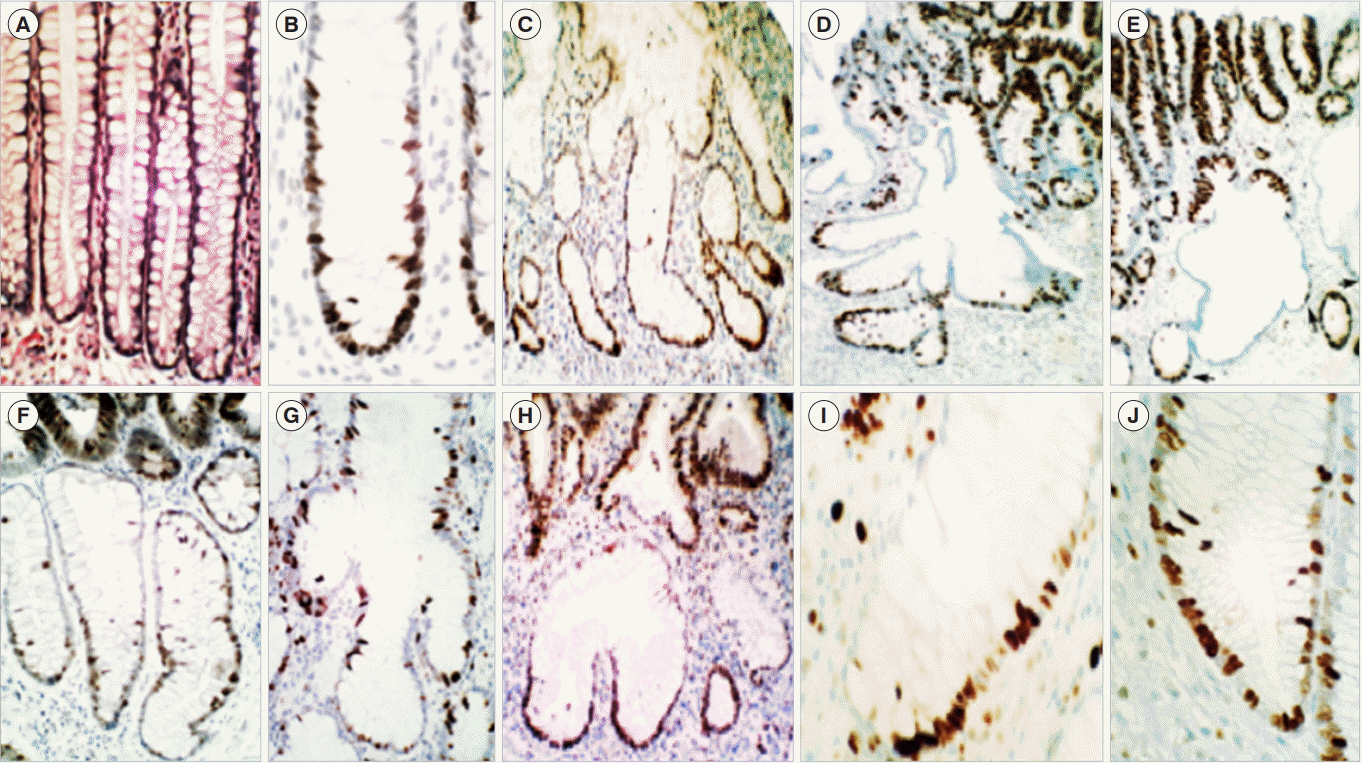
When sections from 60 sporadic polypoid conventional adenomas were immune-stained with the proliferation marker Ki-67, different abnormal cell-proliferation phenotypes were found in the CCS below the neoplastic tissue [45,46]: (1) haphazardly asymmetrically-distributed single proliferating cell (PC) or PC clusters in 80%; (2) continuous PC domains in one or both slopes of the crypts in 17%; and (3) single, haphazardly distributed PCs in the remaining 3% (Fig. 2) [46]. In contrast, the crypts of 12 normal colon segments (controls) exhibited crypts with normal shapes having symmetrically distributed PC-domains predominantly limited to the lower third portion of the crypts. It should be mentioned that in early observations, Maskens and Deschner [47] found upward shift of the proliferative zone in the normal crypts in patients with colon carcinoma. In contrast Arai and Kino [48] found more recently that the proliferative zone in the crypts was normal in patients with colon cancer.
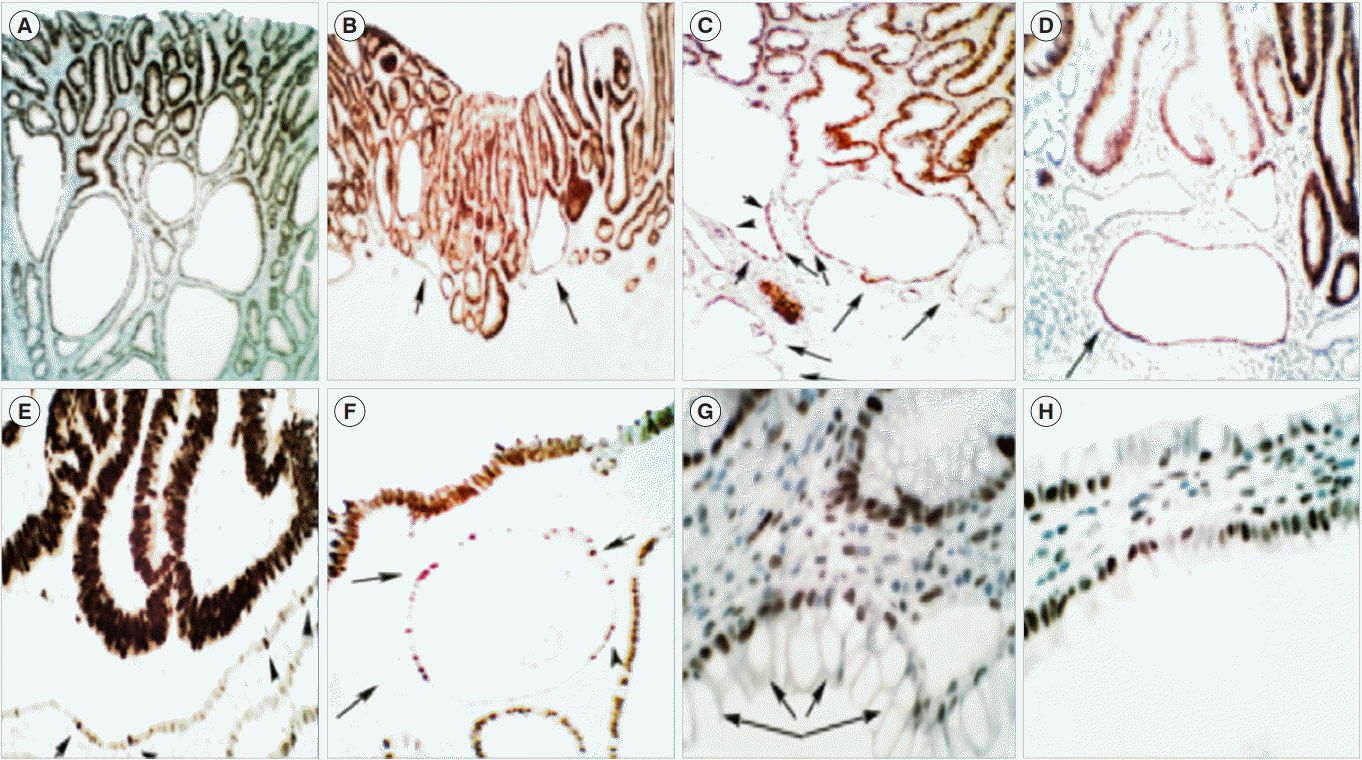
The p53 transcription factor (encoded by the human gene TP53) is a key tumor suppressor that regulates several signaling pathways involved in carcinogenesis. Given the resources required for TP53 gene sequencing, most workers have used p53 immunohistochemistry as a surrogate, assuming that p53 overexpression is connected with a mutation, and that the lack of abnormal expression is indicative of wild type p53 [49]. The immune-stained sections from the 60 sporadic, polypoid conventional adenomas disclosed CCS with haphazardly distributed p53-upregulated, often single cells, in 30% of the sporadic polypoid conventional adenomas (Fig. 3). None of the more than 200,000 crypts investigated in 22 colon segments, proximal or distal to surgically removed colon cancer revealed crypts with p53-upregulated cells [50]. It should be mentioned that in previous studies of colon adenomas we found p53 upregulation in the adenomatous tissue in 28% out of 501 adenomas [51]. Thus, the apparently normal epithelium of the CCS beneath the adenomatous tissue of sporadic polypoid conventional adenomas concurred with an unprecedented relocation of the natural PC domains, and with p53-upregulated cells, suggesting thereby, possible mounting somatic mutations in the CCS of those neoplastic lesions [50].
In another investigation, this time with 48 non-polypoid adenomas, we found that 88% of the CCS exhibited haphazardly asymmetrically-distributed PC-domains, 8% continuous PCdomains in one or both slopes of the CCS, and 4%, asymmetrically-haphazardly distributed single PC [46]. The CCS in 29% of the 48 non-polypoid adenomas also showed p53-upregulated often as single cells [46]. Thus, the CCS accruing beneath the neoplastic tissue of non-polypoid adenomas also thrived with relocated PC-domains and p53 upregulated cells (the latter in nearly one-third of the polypoid adenomas), suggesting cellkinetic alterations and possibly mounting somatic mutations. On the other hand, neither asymmetrically-haphazardly distributed PC-domains, nor p53 upregulated cells were found in any of the more than 200,000 normal colon crypts, including occasional, single CCS in controls.
4. Crypts with CCS in the Mucosa of the Stalk
While studying sporadic polypoid conventional adenomas we detected in the mucosa of the stalk, CCS without neoplastic tissue on top, strongly suggesting that CCS developed independently of the control of the neoplastic tissue [50]. Sections immuno-stained with the proliferation marker Ki-67 and with p53 showed that the CCS in the stalk of sporadic polypoid conventional adenomas displayed haphazardly asymmetricallydistributed PC-domains and p53 upregulated cells. Those unexpected findings suggested that the CCS in the stalk were possibly undergoing somatic mutation [50].
COLON ADENOMAS WITH CORRUPTED CRYPTS IN RODENTS
1. ACF in Rodents
The pristine report of clusters of ACF in the colonic mucosa of carcinogen-treated rodents by Bird [36], was followed by a plethora of publications in rodents, using various carcinogens. A recent search in PubMed (9/30/2019) revealed a total of 1,349 publications on ACF in mice and rats, revealing thereby the interest for the study of ACF as potential markers of CRC risk.
The induction of ACF in rodents is, however, not specific for carcinogen-treated rodents exclusively: Xu et al. [52] found that a high fat diet administration to C57BL mice treated with azoxymethane (AOM) promoted ACF formation concurrent with an increase of opportunistic pathogenic bacteria in the colon of C57BL/6 mice. Moreover, Zhou et al. [53] found that mice fed with sodium nitrate developed ACF. On the other hand, Kristiansen [54] submitted that the presence of a high number of ACF with high crypt multiplicity was predictive for tumor outcome in mice, and Ochiai et al. [41] claimed that the presence of dysplastic ACF facilitated the identification of the early stages of colon carcinogenesis in AOM-treated rats. In contrast, Ghirardi et al. [55] working with Fischer 344 rats, found that ACF were prevalent in the mid colon with lower frequencies in the distal colon and rectum, and rare in the proximal colon and cecum. In their study, ACF growing features and distribution were not correlated to adenoma and adenocarcinoma distribution. The authors concluded that the preneoplastic role of ACF and their function as intermediate biomarkers in colorectal carcinogenesis remained to be clarified [55]. Perhaps, as with humans (see above) it may be summarized that the significance of ACF as potential markers of CRC risk in laboratory animals, remains unsettled.
2. Crypts with Normal Epithelium Albeit CCS in Carcinogen-Treated Rats
As in the human counterpart, the vast majority of the colonic crypts in untreated mice [56] and rats [57] displays close-packed test tube-shaped crypts with straight axes, vertical to the muscularis mucosae. Colon crypts replicate by symmetric fission starting at their base, and proceeding upwards, finally generate 2 separate identical crypts [58]. In a more recent survey we found in the normal mucosa of the colon of a cohort of SpragueDawley rats [59] and Fisher-344 rats [7]–without CRC, despite protracted treatment with the colonotropic carcinogen 1,2 dimethylhydrazine (DMH)–occasional crypts with architectural distortions (CCS). CCS in rats were regarded those with either asymmetric basal fission, asymmetric lateral sprouting/lateral fission, basal dilatations, or spatial aberrations of the normal (vertical) axis. The study of entire Swiss-roll sections from 25 male Sprague-Dawley rats treated with DMH suspended in ethylenediaminetetraacetic acid (EDTA) solution, disclosed a total of 202 CCS (38%) amongst 533 crypts with fission. In contrast, entire Swiss-roll sections from 10 EDTA-treated controls Sprague-Dawley rats exhibited only one CCS amongst the 571 crypts with fission (0.1%). In entire Swiss-roll sections from 25 male Sprague-Dawley rats treated with DMH, 4 adenomas with CCS accruing underneath were found [59]. Hence, despite repeated injections with the colonotropic carcinogen DMH, only occasional adenomas developed limited mucosal hubs, while the vast majority of the remaining colonic mucosa remained histologically unaltered.
In another study 25 male Fisher-344 rats were treated for 24 months, with pellets containing 500 ppm of the mutagenic pyrolysate compound 2-amino-6-methyldipyrido imidazole (GluP-1) [7]. Swiss-roll sections from the entire colon showed that out of 357 crypts with fission (mean number of crypts with fission per animal: 14.2), 130 CCS were found intercalated with normal crypts (mean number of CCS per animal: 5.2; range, 2–12). Three colon adenomas included in the Swiss-roll sections of Fisher-344 rats revealed CCS accruing beneath the adenomatous tissue [7], thus resembling the CCS found beneath sporadic polypoid and non-polypoid adenomas in humans. It was advanced, that the accumulation of CCS in of carcinogentreated rats could reflect mucosal sites from which adenomas ultimately developed, highlighting thereby the first histologic event of “field of cancerization” in the colon of rats.
CONCLUSIONS
In this review, we highlight the occurrence of 2 intertwined compartments coexisting in sporadic conventional polypoid colon adenomas: one consisting of neoplastic (dysplastic) tissue on top, and the other of non-dysplastic crypts with asymmetric, irregular, CCS below. The absence of neoplastic tissue on top of the CCS found in the stalk of those adenomas, should rationally rule out the possibility that the adenomatous tissue had directly orchestrated the growth of crypts with CCS. CCS, both beneath adenomas and in the stalk, coexisted with haphazardly distributed, asymmetric PC-domains. In contrast, none of more than 200,000 normal colon crypts revealed asymmetrically-distributed PC-domains. The rearrangement of PC-domains in CCS beneath adenomas may be the result of the relocation of the cells that fuel the production of PCs, namely the stem cells [2,60]. In addition, p53-upregulated cells were found in the CCS in nearly one-third of the polypoid adenomas suggesting somatic mutations in those crypts. CCS were also found below adenomas in carcinogen-treated rats. The findings that crypts with disparate shapes (CCS) albeit with normal epithelium gather underneath the neoplastic tissue and in the mucosa of the stalk, might open new vistas to study the true histogenesis of conventional adenomas, the most frequent precancerous lesion in the human colon.
 XML Download
XML Download