

 PDF
PDF Citation
Citation Print
Print



INTRODUCTION
Behçet’s disease (BD) is a chronic, relapsing, immune-mediated multisystemic disease characterized by underlying vasculitis [1,2]. BD is particularly more prevalent in the Far East and Mediterranean basin, corresponding to the Old Silk Road, an ancient trading route stretching between the Mediterranean, the Middle East, and the Far East [3] with an estimated prevalence of 13.5–20 cases per 100,000 people in Japan, Korea, China, Iran, and Saudi Arabia [2]. In Japan, the estimated prevalence of BD was reported as 14.9 per 100,000 people in 2002 [4]. According to the diagnostic criteria published by the Japanese Ministry of Health, Labour and Welfare [5,6], the major symptoms of BD include recurrent oral and genital ulceration, skin lesions such as erythema nodosum, subcutaneous thrombophlebitis, pseudofolliculitis and acneiform nodules, and ocular symptoms (iridocyclitis, uveoretinitis). Minor symptoms include arthritis without associated deformity or rigidity; epididymitis; gastrointestinal (GI) symptoms commonly affecting the ileocecal region; vascular lesions in the arteriovenous system and pulmonary vasculature; and moderate or severe central nervous system lesions. Typically, BD is classified into complete, incomplete, or suspected types based on the number of major and minor symptoms, partial or complete appearance of symptoms, and the recurrence or exacerbation of symptoms [5,6].
Specialized categories of BD include vascular, neuronal, and intestinal types. The prevalence of GI involvement causing intestinal ulcers has been reported to be 15%–20% [4], although it varies between different populations [7-12]. Notably, GI involvement is common in Japanese patients with BD and is estimated to be approximately 50% [13,14] and based on an analysis from 2007, has been steadily rising [15]. According to the revised Japanese consensus guidelines (2014) [16], a diagnosis of intestinal BD is based on the presence of a typical oval-shaped large ulcer in the terminal ileum, or ulcerations or inflammation in the small or large intestine. Disease severity is assessed by systemic symptoms (e.g., fever and extraintestinal manifestations), clinical signs of pain, inflammatory mass, and rebound tenderness upon abdominal palpation, depth of ulcers as assessed by radiology and endoscopy, intestinal complications (e.g., bleeding, stricture, and fistula), presence of anemia, and increased levels of inflammatory markers, including C-reactive protein (CRP), white blood cell count, and erythrocyte sedimentation rate. Intestinal lesions generally appear late in the disease course of BD [15], with an age of onset between 18 and 40 years [1]. Intestinal BD is frequently associated with serious complications, such as stricture, perforation, large abscesses, and significant GI bleeding requiring surgical intervention [16], all of which can cause irreversible bowel damage and can potentially be fatal [15].
The main goal of treatment in patients with intestinal BD is to induce and maintain remission in order to minimize disease recurrence, surgical intervention, and the risk of irreversible bowel damage [17]. In the absence of a standard treatment, intestinal BD has been treated with corticosteroids or non-biologic immunomodulators [18,19]; however, many patients experience disease relapse following medical treatment with such agents, with results from a retrospective review in 93 patients with BD reporting cumulative recurrence rates of 24.9% at 2 years and 43.0% at 5 years, despite an initial high remission rate (66.7%) [20]. Moreover, despite their widespread use, there remains a lack of evidence from prospective studies supporting the clinical efficacy of corticosteroids in intestinal BD [21], with only a limited number of case reports demonstrating beneficial effects [22-24]. In addition, patients can become corticosteroid dependent, with a retrospective cohort study of 54 patients with moderate to severe intestinal BD showing that more than one-third of patients (35.2%) demonstrated corticosteroid dependency after 1 year [25]. The revised Japanese consensus guidelines from 2014 recommend induction therapy with prednisolone 0.5–1 mg/kg per day for 1–2 weeks; following clinical improvement, prednisolone should be tapered by 5 mg every week, and then discontinued [16].
The development of the anti-tumor necrosis factor α (TNF-α) class of drugs has improved treatment outcomes in patients with intestinal BD [21]. One such anti-TNF-α, adalimumab (HUMIRA; AbbVie Inc., North Chicago, IL, USA) [26], was approved for the treatment of patients with intestinal BD in Japan in May 2013 [27] based on the results of a 52-week, phase 3 study in 20 Japanese patients with intestinal BD who were refractory to corticosteroid and/or immunomodulatory therapies [12]. In this study, 4 patients (20%) achieved complete remission (score of 0 on global GI and endoscopic assessment; composite efficacy index) and 12 patients (60%) reported a marked improvement (score of ≤ 1 on composite efficacy index) [12]. During the study, 9 of 13 patients (69%) taking corticosteroids at baseline were able to taper (1 patient) or completely discontinue corticosteroids (8 patients). No new safety signals were observed and no deaths occurred [12]. Furthermore, long-term treatment of up to 100 weeks with adalimumab led to a sustained reduction in clinical and endoscopic disease activity, with 40% of patients achieving a marked improvement [6]. In addition, adalimumab was well tolerated with no unexpected adverse events (AEs). The revised Japanese consensus guidelines recommend subcutaneous adalimumab as standard therapy in patients with intestinal BD who have severe disease symptoms with deep ulcers confirmed by radiology or endoscopy. The recommended initial dose of adalimumab in patients with intestinal BD is 160 mg, followed by 80 mg at 2 weeks and 40 mg every 2 weeks thereafter [16].
Since intestinal BD is a relatively rare disorder, evidence from large studies demonstrating the safety and effectiveness of antiTNF-α agents in patients with intestinal BD remains scarce [28]. Consequently, the objective of this study was to evaluate the long-term safety and effectiveness of adalimumab in Japanese patients with intestinal BD through the collection of real-world practice data as part of a pharmacovigilance plan.
METHODS
1. Study Design
This was a prospective, all-case, post-marketing surveillance (PMS) study conducted between 2013 and 2017 at 254 centers in Japan that specialized in medical care for patients with intestinal BD. All centers were equipped to diagnose and manage tuberculosis (TB) and serious infections. As it was planned to analyze the data collected until August 2016, the cutoffs for observation periods were set. Therefore, the observation period varied among patients and was dependent on the time of initiation of adalimumab treatment: 156 weeks when initiated between May 16, 2013, and August 15, 2013; 104 weeks when initiated between August 16, 2013, and August 15, 2014; and 52 weeks when initiated between August 16, 2014, and August 15, 2015. Data for assessment were collected from patient case report forms.
The study protocol was approved by the Pharmaceuticals and Medical Devices Agency, Japan, prior to study initiation. This study was conducted at registered medical institutions in compliance with Good Post-marketing Study Practice (GPSP) in Japan and is registered on ClinicalTrials.gov (NCT01960-790). Approval of the institutional review board of each participating institution and written informed consent were not required because this study was conducted as a regulatory and legal requirement in accordance with the GPSP guidelines of Japan.
2. Eligibility Criteria, Study Assessments, and Endpoints
All patients with intestinal BD in Japan, who received adalimumab following its approval for this indication were included in this study. Adalimumab was administered subcutaneously at an initial dose of 160 mg, followed by 80 mg at 2 weeks and then 40 mg every 2 weeks.
The primary endpoint was the incidence of adverse drug reactions (ADRs). ADRs of interest included infections, TB, malignancy, injection site reactions, autoimmune disease, pancytopenia, demyelinating disease, cardiac failure congestive, and interstitial pneumonia. Secondary endpoints included global improvement rating at study end, the evaluation of global GI symptoms and non-GI BD symptoms in patients reporting these symptoms at baseline, and the degree of improvement in endoscopic findings and CRP levels. Further post hoc analyses were conducted to evaluate baseline factors that may affect the incidence of ADRs.
3. Safety Assessment
The safety population included all patients with a fixed case report form except patients who did not receive the study drug or did not receive it within the study period, patients who reported a protocol violation, patients who were duplicate cases, or those transferred to other hospitals. Safety was evaluated during the study and until 28 days after the observation period. For patients who discontinued adalimumab treatment prematurely and withdrew from the study, the time of the final adalimumab administration was considered as study end and safety was evaluated until 28 days after this final adalimumab administration. ADRs were defined as AEs for which the causal relationship with study drug could not be excluded.
4. Effectiveness Assessment
The effectiveness population included all patients from the safety population except those who were administered adalimumab off-label and for whom a global improvement rating was not available. Global improvement was rated as markedly effective, effective, or ineffective at the last observation time point at the physician’s discretion. Global GI symptoms were scored at baseline, weeks 4, 8, 12, 24, 52, 76, 104, 128, and 156, and at treatment discontinuation based on the following scores: 0, free of symptoms; 1, did not affect a patient’s daily life; 2, slightly affected a patient’s daily life; 3, affected a patient’s daily life; and 4, critically affected a patient’s life. Endoscopic improvement was based on an evaluation of the change in the size of the largest ulcer from weeks 0–24, 24–52, 52–104, and 104–156, and defined as follows: indeterminate, expanded, < 1/4 to no reduction, ≥ 1/4 to < 1/2 reduction, or ≥ 1/2 reduction. Non-GI BD symptoms were assessed at baseline and weeks 52, 104, and 156. CRP levels were measured at baseline and weeks 24, 52, 76, 104, 128, and 156.
5. Statistical Analyses
Quantitative variables were summarized using descriptive statistics (number, mean, standard deviation [SD], median, range). AEs and ADRs were coded using the Medical Dictionary for Regulatory Activities (MedDRA) version 19.1 and summarized as events/100 patient-years (PY) of adalimumab exposure and number and proportion of patients with AEs/ADRs in the safety population. Each safety event was counted as a separate event, even if it occurred multiple times in a single patient. Effectiveness data were collected as observed unless indicated otherwise. The Wilcoxon signed-rank test was used to analyze the global GI score. The impact of baseline factors on the incidence of ADRs was assessed by stratified group analyses and multivariate regression analysis. All tests were performed with a two-sided significance level of 5%.
RESULTS
1. Patient Disposition, Treatment, Demographics, and Baseline Characteristics
Overall, 473 patients were registered in the study, including 462 in the safety and 383 in the effectiveness population (Fig. 1). The safety and effectiveness populations were administered adalimumab for mean (SD) durations of 515.3 (322.2) and 579.5 (307.9) days, respectively. During the study, 395 patients (85.5%) were administered adalimumab per the recommended dosing regimen.
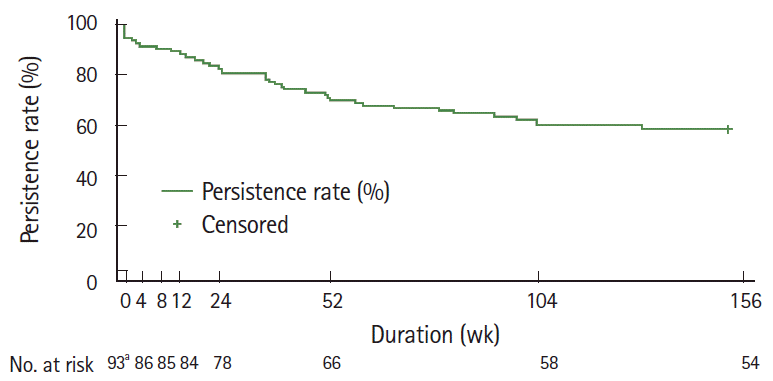
In the safety population, 153 patients (33.1%) discontinued treatment; of these, 142 patients (92.8%) discontinued for known reasons, including inadequate response (33.1%), AEs (29.6%), loss to follow-up (26.1%), patient request (5.6%), and others (5.6%). The treatment persistence rate was 58.1% (54/93; 95% confidence interval [CI], 47.4–67.3) for those patients with an observation period of 156 weeks (Fig. 2).
Patient demographics and baseline characteristics of the safety population are described in Table 1. Overall, 225 patients (48.7%) were female, with a mean age of 46.3 years. In total, 258 patients (55.8%) were diagnosed with complete (25 patients, 5.4%) or incomplete (233 patients, 50.4%) BD. At baseline, 260 patients (56.3%) reported comorbidities, including liver (35 patients, 13.5%) and renal disorders (10 patients, 3.8%). Overall, 445 patients (96.3%) were taking concomitant medications. Self-administration errors were reported in 2 of the 351 patients (0.6%) who self-administered adalimumab.
2. Safety
Overall, the safety population was exposed to the study drug for 689.6 PY. In total, 212 ADRs were reported in 120 patients (25.97%) and 72 serious ADRs were reported in 51 patients (11.04%) (Table 2). The most common ADRs and serious ADRs by MedDRA system organ class (SOC) were “infections and infestations” (any, 10.17%; serious, 3.90%), “investigations” (any, 4.76%; serious, 1.08%), “GI disorders” (any, 4.33%; serious, 2.16%), and “general disorders and administration site conditions” (any, 4.11%; serious, 1.30%). Among ADRs of interest, infections were reported in 47 patients (10.17%), TB in 3 patients (0.65%), injection site reactions in 5 patients (1.08%), and malignancy in 1 patient (0.22%). The incidences of AEs and serious AEs were 46.98/100 PY and 15.37/100 PY, respectively.
Three patients died during the study period, 2 due to a serious infection and one due to malignancy. A male patient who was diagnosed with pneumonia, colon perforation, and an abscess on day 78 of adalimumab treatment; discontinued adalimumab; and was treated with glycopeptide antibiotics and antitrichomonal agents; and was transferred to another hospital where he died. A causal relationship between adalimumab and this serious infection was indeterminable. An elderly woman with a urinary tract infection was admitted to hospital 23 days prior to the start of treatment with adalimumab and then administered adalimumab continuously; however, she developed a catheter-related urinary tract infection on day 15 and sepsis on day 16 resulting in death. Importantly, it was concluded that there was no causal relationship between adalimumab and sepsis. The third patient, an elderly female previously treated for chronic myelomonocytic leukemia (CML) was diagnosed with recurrent leukemia on day 280 of adalimumab treatment and discontinued adalimumab. On day 354, CML recurred and the patient died of increased white blood cell count. A causal relationship between adalimumab and CML could not be excluded.
Two female patients tested positive for TB at baseline screening. The first patient received prophylactic isoniazid 300 mg/day prior to adalimumab initiation. She was diagnosed with pulmonary TB on day 379 of adalimumab treatment and administered antitubercular treatment (ATT; rifampicin 450 mg/day, ethambutol hydrochloride 750 mg/day, and pyrazinamide 1 g/day) and injectable quinolone antibiotics and recovered on day 794. It was concluded that there was a causal relationship between adalimumab and pulmonary TB. The second patient did not receive prophylactic ATT prior to adalimumab initiation and was diagnosed with latent TB again on day 173 of adalimumab treatment following a positive QuantiFERON test. Adalimumab was discontinued and treatment with prophylactic isoniazid 300 mg/day was initiated. However, adalimumab was restarted due to the development of stomatitis, and 15 days later, the patient recovered. A causal relationship between adalimumab and latent TB could not be excluded in this patient. In addition, a male patient, who tested negative for TB at baseline screening, developed TB on day 952 of adalimumab treatment and was transferred to another hospital. The outcome of this patient is unknown; however, a causal relationship between adalimumab and latent TB could not be excluded. One female patient was diagnosed with early-stage breast cancer on day 478 of adalimumab treatment and discontinued therapy. The patient recovered following surgery and causal relationship was excluded.
Of 5 cases with injection site reactions, including one with injection site erythema, 4 cases resolved or were resolving and did so in a mean number of 19.3 days (median, 14 days; range, 1–43 days). The outcome of one case was unknown. No cases of de novo or reactivation of hepatitis B infection were reported.
3. Baseline Factors Affecting Safety
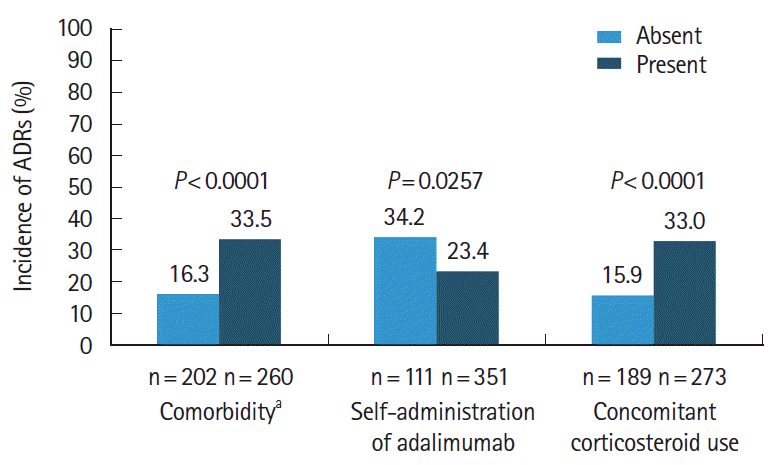
The incidence of ADRs was significantly higher in patients with comorbidities than in patients without comorbidities (33.5% vs. 16.3%; P<0.0001), and in patients taking concomitant oral corticosteroids than in those not taking concomitant oral corticosteroids (33.0% vs. 15.9%; P<0.0001) (Fig. 3). Similarly, multivariate regression analysis demonstrated lower odds of ADRs in patients who had no comorbidities at baseline (odds ratio [OR], 0.4056; 95% CI, 0.226–0.729) and in patients who were not taking concomitant corticosteroids at baseline (OR, 0.5183; 95% CI, 0.292–0.921) (see Supplementary Table 1). The incidence of ADRs was significantly lower in patients who self-administered adalimumab compared to those not self-administering adalimumab (23.4% vs. 34.2%; P=0.0257), with multivariate regression analysis demonstrating higher odds of ADRs in patients who did not self-administer adalimumab (OR, 1.8897; 95% CI, 1.011–3.532) (see Supplementary Table 1).
An assessment of the impact of each of these patient background factors (comorbidities, concomitant oral corticosteroid use, and self-administration of adalimumab) in relation to the onset of the 4 most commonly reported ADRs by MedDRA SOC (“infections and infestations,” “GI disorders,” “general disorders and administration site reactions,” and “investigations”) is shown in Table 3. Overall, serious ADRs were more frequently reported in patients who were not self-administering adalimumab compared to those who self-administered adalimumab, a finding that was observed across the 4 most common ADRs by MedDRA SOC (“infection and infestations,” 8.11% vs. 2.56%; “GI disorders,” 4.50% vs. 1.42%; “general disorders and administration site reactions,” 2.70% vs. 0.85%; and “investigations,” 3.60% vs. 0.28%). Notably, in patients receiving concomitant corticosteroids, the incidence of any and serious infections and infestations was higher (any, 13.92%; serious, 5.13%) compared to those who were not receiving concomitant corticosteroids (any, 4.76%; serious, 2.12%).
4. Effectiveness
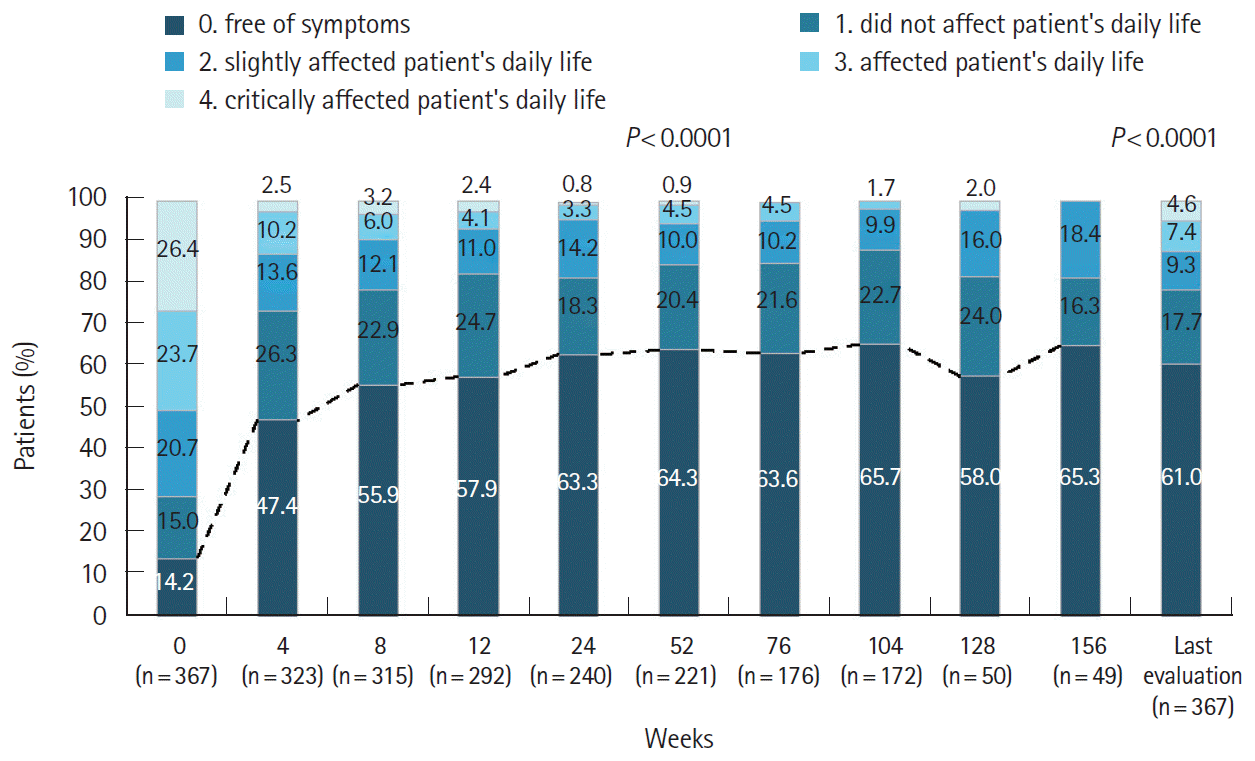
In the effectiveness population, 324 patients (84.6%) had a global improvement rating of “effective” (n = 156, 40.7%) or “markedly effective” (n = 168, 43.9%) at study end. The percentage of patients with a global GI score of 0 increased from 14.2% at baseline, further increasing to 63.3% at week 24; these improvements were maintained over the course of the study (Fig. 4). Likewise, the percentage of patients with a global GI score of 1 increased from 15.0% at baseline to 26.3% at week 4 and was sustained thereafter (Fig. 4). There was also a significant (P<0.0001) increase in the percentage of patients with an improved global GI score at week 52 and at the last evaluation time point.
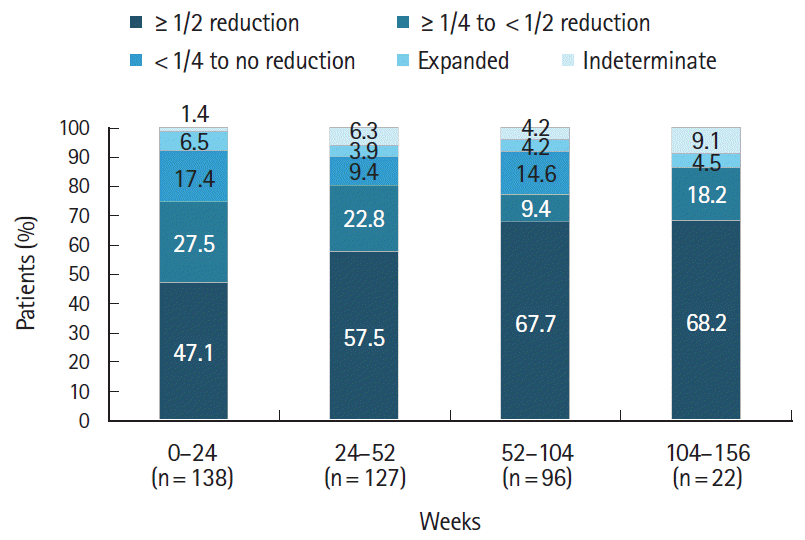
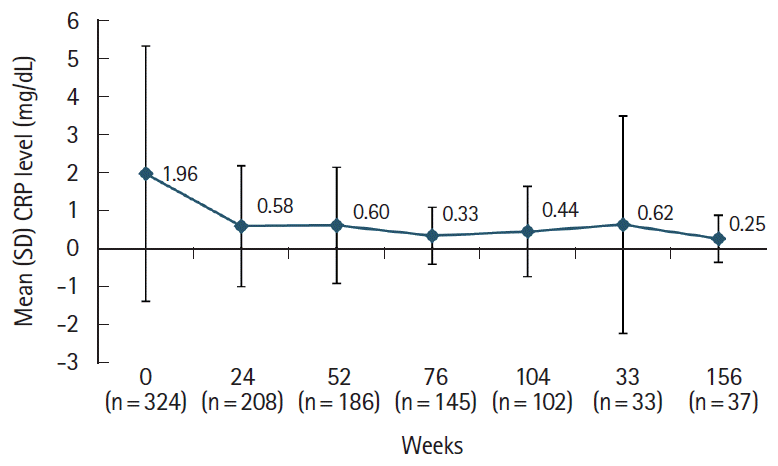
Upon endoscopic evaluation, the percentage of patients with a ≥ 50% reduction in the size of the largest ulcer gradually increased from 47.1% at weeks 0–24 to 68.2% at weeks 104–156 (Fig. 5). Treatment with adalimumab was also associated with resolution of non-GI BD symptoms. From baseline to the last evaluation time point, there was a decrease in the number of patients reporting oral aphthous ulcers (183 vs. 62 [33.9%]), skin symptoms (96 vs. 23 [24%]), eye symptoms (4 vs. 0 [0%]), and genital ulcers (51 vs. 13 [25.5%]) (Table 4). Following adalimumab treatment, mean ± SD CRP levels decreased more than 3-fold from baseline (1.96 ± 3.36 mg/dL; n = 324) to week 24 (0.58 ± 1.59 mg/dL; n = 208). Further gradual decreases were observed up to week 156 (0.25 ± 0.62 mg/dL; n = 37) (Fig. 6).
DISCUSSION
To the best of our knowledge, this is the first study to evaluate the long-term safety and effectiveness of an anti-TNF-α agent in a large population of patients with intestinal BD. Notably, nearly 60% of patients who were followed up for 156 weeks continued treatment, indicating that adalimumab was well tolerated over the long-term. Overall, the safety profile of adalimumab was consistent with that previously reported in clinical trials in patients with BD in Japan [6,12], as well as in other approved indications [29]. Moreover, this study did not identify any new safety signals.
The most frequently reported ADRs were “infections and infestations,” “GI disorders,” and “general disorders and administration site conditions;” these findings were consistent with those from a recent 24-week PMS that evaluated the safety and effectiveness of adalimumab in 1,693 Japanese patients with Crohn’s disease (CD) [26]. Crucially, the incidences of ADRs (30.74/100 PY) and serious ADRs (10.44/100 PY) reported in this study were lower than those reported in the PMS study in patients with CD (ADRs, 76.1/100 PY; serious ADRs, 18.3/100 PY) [26], highlighting that long-term administration of adalimumab in a real-world setting was well tolerated. In addition, the rate of serious infections reported in this study (3.92/100 PY) was consistent with that previously reported in other disease states following adalimumab treatment (4.7/100 PY and 5.1/100 PY in 2 rheumatoid arthritis studies and 4.5/100 PY and 6.6/100 PY in 2 CD studies) [26,29-31].
There is a recognized risk of TB during anti-TNF-α therapy [29,30]. Consequently, TB was an ADR of particular interest in this study. Overall, the incidence of TB of 0.15/100 PY in this study was comparable to that of a long-term safety analysis in 15,132 patients receiving adalimumab in global rheumatoid arthritis trials [29]. In total, there were 3 reported cases of TB, including 2 patients who tested positive for TB at baseline and received ATT. Both patients recovered following treatment with ATT and continued adalimumab treatment. All patients require testing for TB and if positive, should receive ATT prior to initiation of adalimumab [30]. However, nearly one in 5 patients in the current study were not screened for TB; this may be explained by the fact that a relatively high proportion of patients had previously received treatment with an anti-TNF-α agent, and were likely tested prior to enrolment into this study.
An assessment of potential patient baseline background factors that may affect the onset of ADRs revealed that the incidence of ADRs was significantly higher in patients with comorbidities, patients taking concomitant oral corticosteroids, and those not self-administering adalimumab. These findings were confirmed by the results of multivariate analyses that demonstrated an approximately 40%–50% reduction in the odds of ADRs in patients without comorbidities and who were not taking concomitant oral corticosteroids, and an approximately 2-fold increase in the odds of ADRs in patients who were not self-administering adalimumab. This is in line with the findings from the PMS study in Japanese patients with CD, which also identified concomitant use of corticosteroids, comorbidities, and injections administered by medical personnel as risk factors affecting the safety of adalimumab treatment [26]. Likewise, results from an analysis of the phase 3, randomized, double-blind, 1-year CHARM trial and its 2 year open-label extension trial ADHERE in patients with CD on adalimumab treatment, reported that the incidence of serious infections was higher in patients taking corticosteroids at baseline than in those who were not [32]. Interestingly, similar findings have been reported with the anti-TNF-α infliximab, with results from the TREAT registry that examined long-term safety data in 6,290 patients with CD reporting that concomitant corticosteroid use was an independent predictor of serious infection (OR, 2.21; 95% CI, 1.46–3.34; P<0.001) [33]. Taken together, these findings highlight an association between concomitant corticosteroid use and the risk of serious infections, highlighting the need for increased clinical awareness of these potential infections, especially early in the course of treatment.
Pathergy reaction, a non-specific, skin hypersensitivity to a needle prick, is a unique feature of BD and also, one of the minor diagnostic criteria by the International Study Group for Behçet’s Disease [34]. However, injection site reactions were reported in only approximately 1% of patients in this study. Moreover, the incidence of injection site reactions in this study (0.73/100 PY) was considerably lower than that of general disorders and administration site reactions (14.3/100 PY) reported in the PMS in Japanese patients with CD [26], potentially suggesting that pathergy does not affect the incidence of injection site reactions in patients with BD receiving adalimumab treatment. Interestingly, self-administration errors were only reported in 2 of 351 patients (0.6%) who self-administered adalimumab. This is considerably lower than that reported from the PEARL survey, which questioned 124 outpatients with CD on adalimumab self-injection at 4 university hospitals, and reported that injection failures, including leakage of drug solution, disconnection of the needle, and the needle failing to reach the subcutaneous areas, occurred in 11.4% of patients. However, the PEARL survey also reported that self-administration was linked to greater patient satisfaction and improved drug adherence [35]. This, combined with the fact that both our study and the PMS study in Japanese patients with CD [26] reported injections administered by medical personnel as risk factors affecting the safety of adalimumab, suggests that self-administration of adalimumab could be safely implemented in patients with intestinal BD. However, it is probable that many of the patients who received injections from medical personnel in this study had more severe disease and were therefore not permitted to self-inject.
This long-term study also confirmed the rapid and consistent rate of disease improvement with adalimumab, which was observed as early as week 4. At week 52 in the current study, 85% of patients were free of symptoms or reported symptoms that did not affect daily life (global GI score ≤ 1), which was slightly higher than that achieved in the phase 3 trial in Japanese patients with BD [12]. Moreover, this percentage increased to 88% at week 104, which was considerably higher than that reported in the open-label extension of the phase 3 trial at week 100 (45%) [6]. These improvements were sustained over the course of the study.
In line with previously reported studies in patients with intestinal BD [6,12], improvement in the endoscopic appearance of ulcers was observed early, with the largest ulcer reduced by ≥ 50% in 47.1% of patients between weeks 0 and 24; this increased to 67.7% of patients between weeks 52 and 104 and was sustained thereafter, thereby confirming the long-term efficacy of adalimumab in patients with intestinal BD in realworld practice. Previous studies reported that adalimumab is associated with the resolution of non-GI BD symptoms, with results from the phase 3 trial in Japanese patients with BD reporting that two-thirds of patients with oral aphthous ulcers and genital ulcers and 88% of patients with erythema nodosum had complete resolution of these symptoms at week 52 [12]. In addition, results from retrospective studies and case series have also reported resolution of both oral ulcers [36] and ocular symptoms [37-39] following adalimumab treatment. These findings have also been replicated in patients with CD, with results from CARE study, a large pan-European cohort of 945 patients with moderate to severe CD [40], and a post hoc analysis of pooled data from 11 adalimumab clinical trials [41] reporting substantial rates of resolution of CD-associated extraintestinal manifestations, including oral, skin, and ocular symptoms, following adalimumab treatment. Unsurprisingly, treatment with adalimumab was also associated with resolution of nonGI BD symptoms in this study. Overall, two-thirds of patients with oral aphthous ulcers and three-quarters of patients with genital ulcers and skin symptoms had resolution of these conditions at the last evaluation time point. In addition, all patients achieved resolution of ocular symptoms at the last evaluation time point. The mean serum levels of CRP decreased from baseline following adalimumab treatment, demonstrating biologic anti-inflammatory activity.
During the course of the study, just under one-third of patients were able to discontinue corticosteroids at week 52. This was considerably lower than that reported in the phase 3 trial and its open-label extension in Japanese patients with BD, where two-thirds of patients discontinued corticosteroid use at week 52 and over 50% of patients discontinued at week 100 [6,12]. However, in the phase 3 study, investigators were permitted to taper and discontinue corticosteroids in responders from 8 weeks and 24 weeks onwards, respectively [12]. In contrast, there were no such predefined criteria in the current study, with corticosteroid discontinuation based on the treating physician’s discretion; this factor could have contributed to the lower proportion of patients who discontinued corticosteroids in the current study. Moreover, baseline data were not available for all baseline corticosteroid users at all time points. Nevertheless, results from this study highlight that patients can successfully discontinue corticosteroids in real-world practice.
This study has several strengths. Of particular importance is the fact that the safety and effectiveness of adalimumab in a large population of patients with intestinal BD could be assessed over the long-term in routine clinical practice. This study also provides further evidence on the use of anti-TNF-α treatment in patients with intestinal BD, which is a rare form of BD, in a real-world setting. As this study enrolled patients with intestinal BD who received adalimumab following its approval for this indication and without any additional inclusion criteria, results can be generalized to the population of Japan with intestinal BD. However, this study has several limitations. Some of the limitations are inherent to the observational design of the PMS, including potential bias that may arise from open-label evaluation. In addition, routine clinical settings resulted in limitations such as the inability to control use of concomitant treatment and the inability to obtain data at all time points, which may compromise interpretation of data. In conclusion, results from this real-world study in a large cohort of patients from Japan demonstrated that adalimumab is safe and effective for the treatment of intestinal BD. Long-term treatment with adalimumab was well tolerated without any unexpected AEs. In addition, adalimumab treatment led to sustained reduction in clinical and endoscopic disease activity.

 XML Download
XML Download