

 PDF
PDF Citation
Citation Print
Print



INTRODUCTION
Inflammatory bowel disease (IBD) is an intestinal inflammatory condition that affects more than 2 million and 0.2 million people in the United States and Japan, respectively. Initially, the 3 major genetically manipulated mouse models of spontaneous intestinal inflammation including interleukin-10 knockout (IL-10 KO), IL-2 KO and T cell receptor alpha chain (TCRα) KO mice, were established in 1993 [1-3]. Since then, experimental murine models of intestinal inflammation that mimic IBD have become increasingly common over the last 2.5 decades. These include experimentally induced and genetically altered models as well as spontaneously developed intestinal inflammatory models [4-6]. Currently, so many different kinds of spontaneous IBD animal models, which were generated by genetically engineered technologies, have emphasized the presence of extremely complicated imbalance of internal/external factors during the development of IBD [4-7]. In particular, a disturbance to gut microbiota homeostasis, so called dysbiosis, has been implicated a wide range of diseases including not only IBD but also obesity, allergic disorders, diabetes mellitus, autism spectrum disorders, and colon cancer [7-9]. Therefore, dysbiosis should be always taken into consideration when we discuss the complicated pathogenesis of several disorders. Various factors including genetic factors, immunological factors, environmental factors, and diet are highly involved in the formation of gut dysbiosis [7]. To closely understand the interplay of the above factors, animal models of IBD are indispensable. So far, at least 66 different kinds of animal models of IBD have been established, and these are classified into chemically induced, adaptive cell transfer, congenital mutant and genetically engineered models [10]. Furthermore, recent advances on genetically engineered techniques allowed us to utilize cell-specific/inducible KO as well as knock-in mouse systems in IBD studies [4,10].
Based on the result of human genome-wide association studies (GWASs), several IBD susceptibility genes have been identified so far [11-14]. In particular, IL-10 receptor and its signaling components seem to be very critical in regulating intestinal homeostasis and immunity, and polymorphisms of those genes in innate immune cells are associated with dysregulation of mucosal immune tolerance and development of early onset of IBD [14-16]. To analyze animal models that manipulate the IBD susceptibility genes may strongly enhance our understanding of their roles in IBD pathogenesis. Furthermore, experimental models of IBD have provided a way to closely dissect mechanisms of understanding the pathogenesis of IBD, which will be associated with developing multiple and critical inventions for IBD therapeutic strategies (e.g., small molecules, biologics) currently.
MOUSE MODELS OF IBD
Currently available mouse models of IBD have been divided into the following 4 categories as shown below.
1. Genetically Engineered Murine Models of IBD
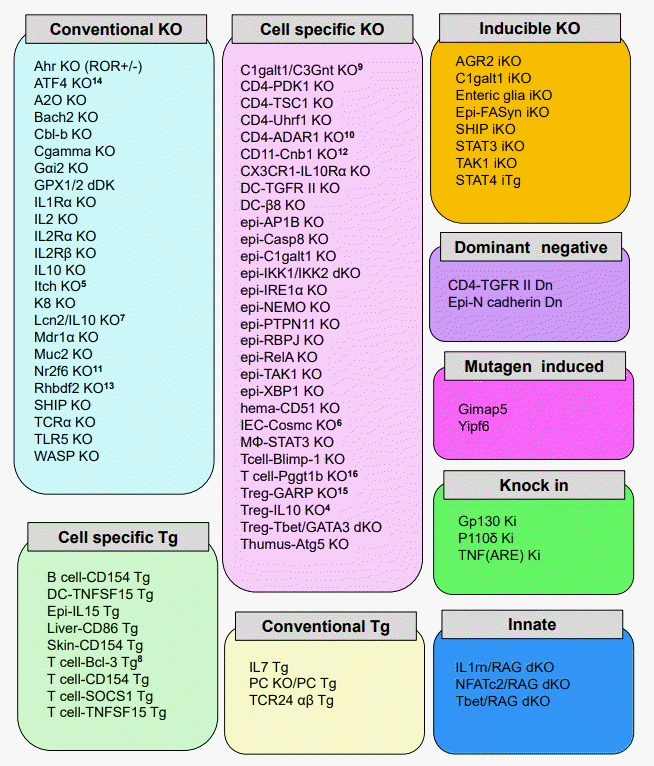
Recent advances on genetic engineering technologies allowed us to analyze the influences of overexpression or deficiency of particularly interesting genes during the development of IBD. Based on the gene construction strategies, genetically engineered murine models of IBD are classified into 9 groups, which includes conventional transgenic (Tg) or KO mice models, dominant negative Tg models, cell-specific Tg or KO models, inducible KO models, knock-in models, innate models, and mutagen-induced models (Fig. 1). In particular, conventional KO mice, which are genetically engineered to lack a particular gene of interest in all cell types, are commonly used to study the pathogenic/regulatory factors during the development of spontaneous chronic colitis [1-4]. It is dispensable to utilize genetically engineered mouse models of IBD for understanding the pathogenesis of IBD and these models have been providing important insights into the pathogenic mechanisms of chronic colitis development. Although the intestinal inflammation seen in these models does not exactly replicate human IBD and cannot be a complete surrogate for the human disease, some characteristically pathological features are relatively reproducible and open for investigation [17].
Cell-specific KO (conditional KO) mouse is made by crossing the flox mouse (which gene loci sandwiching the target gene region with the Cre recombinase target sequence loxP) and the mouse with expressing tissue/cell-specific Cre recombinase. As a result, a particular target gene can be deleted in tissues/cells that form specific organs. Inducible KO mice can reversibly control the expression of the gene of interest. Conventional Tg mice overexpress all cells by introducing the target gene. In contrast, cell-specific Tg mice can be overexpressed in specific cells and organs by using viral vectors or electroporation (Fig. 1).
Dominant negative Tg mice are genetically engineered to overexpress non-functional proteins that interfere with the normal function of the target protein, whereas knock-in mice have a mutation in the gene of interest including the IBD susceptibility genes. The innate model has a lack of specific genes that cause immunodeficiency. Finally, in the mutagen-induced model, genetic variation is induced by N-ethyl-N-nitrosourea (ENU), a highly potent mutagen. In summary, these genetically engineered mouse strains are very useful for analyzing gene functions in elucidating the complex IBD mechanisms.
2. Chemically Induced IBD Models
Currently, chemically induced colitis models belong to one of the most commonly used mouse IBD models since the onset of inflammation is immediate and the procedure is generally straightforward [18]. Chemically induced colitis models are suitable for dissecting the pathogenic/regulatory components during different phases of colitis, such as acute, recovery and chronic phases [19]. These models enable us to pick up some novel genes that are highly involved in IBD susceptibility, and also to identify some immunological/biological mechanisms during the development of acute/chronic colitis [18,19]. Oral administration of dextran sodium sulfate (DSS), induces colonic epithelial injury and has been widely used as not only acute but also chronic colitis model [19,20], which is useful for assessing mechanism of wound healing process and inflammation of associated epithelial homeostasis. However, chemically induced colitis models need to optimize concentration of chemicals depending animal facilities. In addition, colitis induced by some chemicals by single administration usually shows acute colitis features and self-limiting without any treatment.
One cycle of 3% to 5% DSS administration for 4–7 days, followed by regular water, results in acute injury with partial crypt depletion on the distal segment of colon. The colon shows multiple and skipped mucosal erosive lesions with mixed inflammatory cell infiltration of mononuclear/polymorphonuclear cells [20]. Since DSS termination induces the recovery of colonic mucosa gradually, this colitis model has been frequently used for studying epithelial wound healing during the recovery phase [19]. After the late recovery phase, macrophages and CD4+ T cells become prominent in areas of wound healing in the basal portion of the lamina propria [19]. However, DSS-induced acute colitis can be still induced in the absence of acquired immunity as severe combined immunodeficiency (scid) mice are also susceptible. Therefore, this colitis is a simple model of acute chemical injury rather than chronic inflammation, and researchers should not use it as a model for human UC. Similar to the DSS colitis model, rectal administration of hapten reagent 2,4,6-trinitrobenzene sulfonic acid (TNBS) with ethanol is widely utilized as a model for intestinal inflammation in rat and mouse [21,22]. This TNBS-colitis model has many features of CD in humans, including severe transmural colitis, which is induced by Th1/Th17-mediated immune responses [23]. In contrast, rectal administration of ethanol suspended oxazolone, another haptenating agent, leads to an IL-4 driven Th2-type colitis, which is limited to the distal part of the colon with relatively superficial ulceration of epithelium [24].
In 1978, MacPherson and Pfeiffer [25] found that administration of 3% to 5% acetic acid induces acute colitis with initial epithelial necrosis and edema in rats. Similarly, administration of acetic acid also causes colonic epithelial destruction followed by inflammatory cell infiltration in mice [26]. In contrast, subcutaneous administration of indomethacin, a NSAID, induces acute type of ileitis, in particular distal jejunum and proximal ileum, in rats [27]. In general, chemically induced colitis models are easy to initiate inflammation and are very useful for the screening of new drugs for human IBD.
3. Adaptive Cell Transferred Models
Naïve CD4+ T cell population are included in the CD45RBhigh fraction, and memory CD4+ T cells, which are already primed by specific antigen, are included within CD45RBlow fraction [28]. Powrie et al. [29] reported that adaptive transfer of CD4+ CD45RBhigh T cells, which are isolated from spleen of wild-type mice, into scid mice result in subacute colitis approximately 6 to 12 weeks after cell transfer. However, scid mice that received CD45RBhigh and CD45RBlow fractions together do not develop severe colitis since CD45RBlow fraction includes regulatory T cells [29,30]. This model allows to examine some of the earliest immunological factors involved in the induction and/or perpetuation of colitis [31]. However, CD45RB cell transfer model needs to purify the CD45RBhigh cells by utilizing a cell sorter and the expert intravenous injection skill is required to administer the isolated CD45RBhigh cells to mice.
4. Congenital (Spontaneous Gene Multination) Models
C3H/HeJBir (C3Bir) mice, which have a missense mutation in the third exon of the Tlr4 (Toll-like receptor 4) gene, spontaneously develop inflammation in cecum and colon [32]. The inflammation peaks at 3–6 weeks old with resolution by 12 weeks old with an occasional recurrence of colitis after 1 year old [32]. Interestingly, IL-10-deficient C3Bir mice developed much more severe colitis with severe ulcerative inflammation, epithelial hyperplasia as compared to IL-10 KO mice [33].
The SAMP1/YitFc mouse strain develops CD-like ileitis around 10 weeks of age with elevated interferon-gamma (IFNγ) and TNF-α [34]. Global expansion of the stem cells in crypt epithelium, of which accompanied with severity of colitis, can be observed in these mice [35]. Of note, both IL-13 and IL-5 expression levels are significantly increased in these mice, suggesting the Th2 pathway seems to also contribute during the chronic stage of inflammation [35]. One of the highest advantages of the SAMP1/YitFc mice model is that it can be utilized as a close CD model, which shows perianal disease with fistula formation in approximately 5% of mice. However, this spontaneous ileitis takes more long time (>30 weeks of age) to develop transmural inflammation in the terminal ileum with 100% penetrance [17].
IBD SUSCEPTIBILITY GENES AND THOSE ANIMAL MODELS
Since the identification of IBD susceptibility gene NOD2 (nucleotide-binding oligomerization domain-containing protein 2) in 2001, more and more genes of IBD are being identified by GWASs. In humans, more than 350 genes have now been identified. In animal experiments of IBD, it has been reported that more than 74 types of genetically engineered mouse strains spontaneously develop colitis so far [4]. The IBD susceptibility gene deficient mice models can be utilized to clarify the function and the role of particular genes during the development of intestinal inflammation. However, the phenotype may be different depending on the cell or tissue type in which the IBD susceptibility gene is abrogated.
Nod2 is a CD-specific susceptibility gene and NOD2 recognizes single-stranded RNA of bacteria and viruses, removes foreign substances through the activation of nuclear factor-κB, and is involved in innate immunity [36]. In addition, about 78 genes such as PTPN22, IL2RA, IL27, TNFSF11, and VAMP3 have been identified as CD-specific susceptibility genes so far [4]. In UC, there are about 59 UC-specific susceptibility genes including HNF4A (involved in the intestinal epithelial barrier mechanism) and CDH1 (encoding E-cadherin, a cell adhesion molecule) [4]. More than 140 susceptibility genes common to CD and UC have been reported. In particular, the IL23R/ Th17 signaling pathway of IL-23R, IL-22, IL-10R2, STAT3 (signal transducer and activator of transcription 3) and MUC1 (mucin 1) is especially important for the development of colitis [37,38].
Genetically manipulated IBD animal models including KO and Tg mice of the above human susceptibility genes are utilized to further prove the direct/indirect association(s) of those genes in IBD. Among them, IL-10RA and/or IL-10RB mutations cause very severe early onset IBD due to unregulated regulatory signals via IL-10 [15,16]. In contrast, Nod2 KO mice as well as Atg16L1 mutant mice do not develop spontaneous colitis or ileitis [38,39]. Although Atg16L1 mutant mice do not develop spontaneous intestinal inflammation, these mice do cause Paneth cell dysfunction and endotoxin overproduction [40,41]. These results suggest that a single mutation of Nod2 or Atg16L1 is not sufficient for spontaneous induction of colitis, and the pathogenesis of IBD seems to be more complicated than we initially predicted [42].
PATHOGENIC FACTORS IN ANIMAL MODELS OF IBD
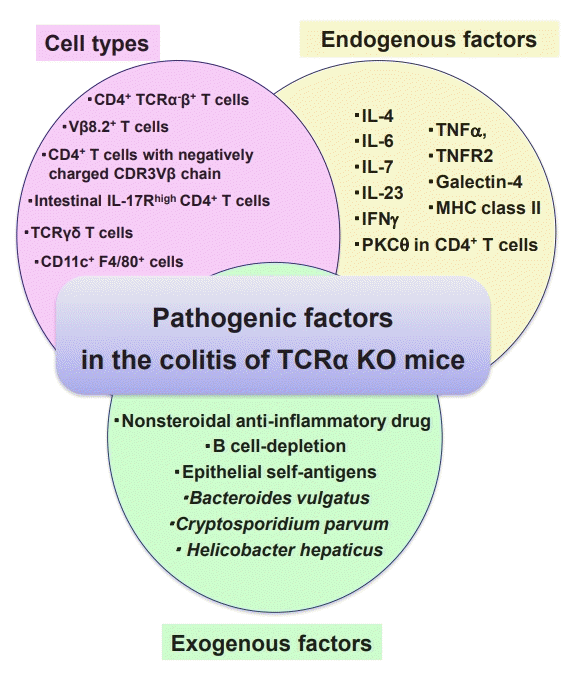
There are many genetically modified mice that spontaneously develop colitis (Fig. 1). Within those models, T cell receptor α chain (TCRα) KO mice established by our group in 1993, have been often used as a murine model of UC and useful for identification of many pathogenic factors involved in IBD [3,43,44]. Approximately 60% of TCRα KO mice develop Th2-mediated colitis by 6 months of age [3,43]. However, the severity of colitis and the frequency of onset of colitis in TCRα deficient mice alter depending on strains and animal facilities [3]. TCRα KO mice have impaired thymic selection (abrogation of negative selection), resulting in the lack of regulatory T cells and differentiation of CD4+ TCRα-β+ T cells, which develop colitis mediated the production of IL-4 [45-47]. These TCRα-β+ T cells are immunologically functional as shown by cytokine profile and the TCR repertoire pattern [46,47]. Interestingly, it has been clarified that these pathogenic TCRα-β+ T cells are also present in human with an autoimmune condition [48].
Under germ-free conditions, the onset of colitis was suppressed in TCRα KO mice [49]. Moreover, TCRα KO mice spontaneously develop colitis independently of Helicobacter species, whereas TCRβ KO mice develop Helicobacter species dependently [50,51]. Furthermore, the permeability of epithelial cells, which is prone to increase in the context of bacterial infection, remains unchanged in TCRα KO mice [17]. Interestingly, the development of colitis in TCRα KO mice was not observed in facility under conventional conditions, and regulatory B-1 B cells seem to prevent the development of colitis under non-hygienic conditions [52,53].
As shown in Fig. 2, several cell types and factors have been identified that play pathogenic roles in the colitis of TCRα KO mice. Another interesting feature of TCRα KO mice is the important role of regulatory B cells, so called Breg, in the colitis [54,55]. In B cell deficient TCRα KO mice, colitis much more worsened as compared to TCRα KO mice, suggesting that B cells play a protective role in colitis [54-56]. Although B cells were previously thought not to be the main source of cytokines, the B cells in colitis TCRα KO mice with colitis produce large amounts of IL-10 and IL-12 (p70), suggesting that they contribute to improvement of the disease [56,57]. These findings suggest the existence of regulatory B cells as well as pathogenic B cells in UC patients. Indeed, results of randomized placebo-controlled trials have been shown that B cell depletion therapy by rituximab (humanized anti-CD20 monoclonal antibody [mAb]) is ineffective in inducing remission of moderately active UC [58]. Furthermore, a case report shows that B cell depletion with the anti-CD20 mAb correlates with IL-10 production and exacerbates UC [59]. In the other 3 case reports, the development of UC patients progressed after B cell depletion therapy and was induced complication of other autoimmune diseases [60].
There are lots of genetically engineered mice, which develop spontaneous colitis (Fig. 1). Pathogenic factors in IBD including dysregulated immune responses, impaired epithelial barrier function, the cytokines, and microbiota could be identified by utilizing these mouse models of IBD.
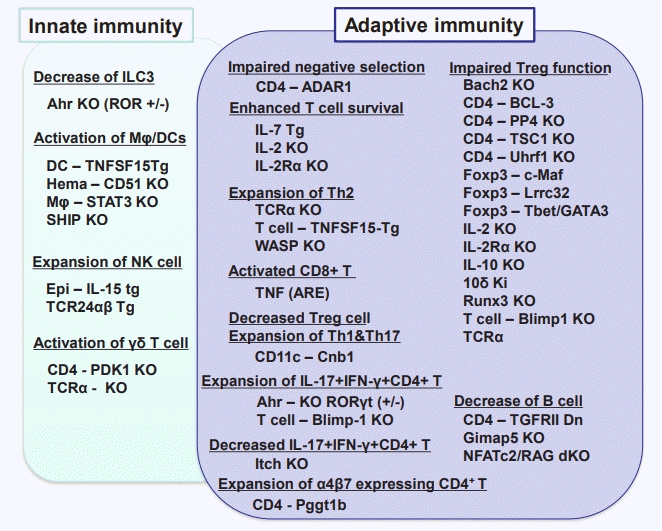
Forty kinds of immune cell-specific KO mice are spontaneously induced the intestinal inflammation, which dues to the reasons such as dysregulated innate and adaptive immunity responses (Fig. 3). In addition, 18 kinds of intestinal epithelial cell-specific KO mice develop spontaneous colitis, which are caused by impaired mucosal barriers, unregulated necroptosis/apoptosis, reduction of antibacterial peptide and/or abnormal response to endoplasmic reticulum stress (Fig. 4).
As shown in Fig. 1 as well as some of the references, IL-10 is one of the most important anti-inflammatories and regulatory cytokines, which is produced by many kinds of cells including macrophages, T cells and B cells [15,16]. Kühn et al. [1] established IL-10 KO mice, which spontaneously develop chronic enterocolitis after 3 months of age with marked regenerative crypt hyperplasia in the colon. Enteric flora plays the key role during the development of colitis in IL-10 KO mice: The inflammation is much milder (restricted in the colon but not in the small intestine) under specific pathogen-free conditions as compared to conventional conditions, and the colitis has been shown to be dependent on Helicobacter-species infection [61]. Furthermore, colitis in IL-10 KO mice is abolished under germ-free condition suggesting resident enteric bacteria are required for spontaneous colitis and immune system activation in IL-10 KO mice [62]. Interestingly, anaerobic bacteria and aerobic bacteria play different role in this colitis: Metronidazole is effective for the treatment of colitis but not typhlitis (inflammation in the cecum), while Ciprofloxacin is beneficial for the treatment of typhlitis but not colitis [63]. Although the colitis in IL-10 KO mice are often referred as Th1-type, IFNγ plays a protective role in this colitis since IFNγ×IL-10 DKO mice develop more severe colitis as compared to IL-10 KO mice [64]. In fact, recent GWAS have identified Il10 as susceptibility gene for both UC and CD, so we should not use IL-10 KO mice as a simple CD-like model [17].
ROLE OF ANIMAL MODELS IN IBD TREATMENT
Currently, several modalities of treatments could be considered for the management of IBD. These include (1) small molecules (e.g., corticosteroids and mesalazine), (2) biologics (e.g., anti-TNF-α), (3) emerging therapies (e.g., stem cell therapy and fecal microbiota transplantation [FMT]), as well as (4) alternative therapies (e.g., traditional Chinese medicine). Animal models could provide a platform for inference of efficacy and safety for the development of some of these treatments and will continue to play a supporting role in facilitating the discovery of new drugs and treatment for IBD.
Interestingly, in one of the highly cited studies on translational of research evidence from animals to human showed that only 10% of these interventions from animal studies were subsequently developed into something that could be fit for patient use [65]. In the case of biologics, there are more than 60 potential new targets involving more than 600 phase I-III clinical trials, but only a few, including anti-TNF-α agents, natalizumab, ustekinumab and vedolizumab, made it to the clinical application eventually. This is likely due in part to poor methodology and failure of the models to accurately mimic the human disease condition and suggested improvements to increase translational success rates have been previously proposed [66].
Certainly, there are limitations inferring observations from animal models. Particularly, human IBD is polygenic and multifactorial in nature, in contrast to experimental IBD animal model that usually involves disruption or overexpression of single genes and thus limiting systemic pathways. Another limitation of extrapolating animal models into human safety and efficacy is the qualitative and quantitative measurement of outcomes. Human clinical trials for IBD measures disease activities through various indexes that quantify symptoms related to active disease, including the CDAI and the Mayo Clinic score for UC. In contrast, efficacies of interventions in animal models are usually assessed through histopathology score of colonic inflammation and clinical score. These parameters might not be sufficient to make inference to clinical remission in heterogeneous groups of patients. In addition, other pharmacological considerations, including bioavailability, time and route of administration, as well as dosing are also important factors to affect the difference between experimental colitis data with actual human context.
The subsection below provides a deeper dive into how animal contributes towards the development of the 2 main modalities of treatment, small molecules, and biologics for IBD.
1. Small Molecules
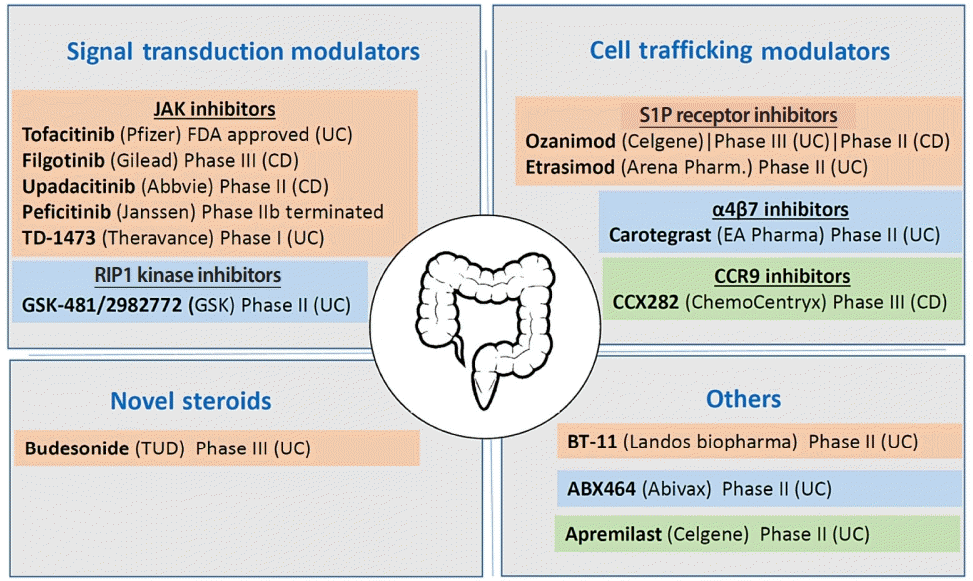
Small molecules medications remain the most common used first line of treatment for IBD. These include 5-aminosalicylates and immunosuppressive agents such as methotrexate and thiopurines, as well as corticosteroids which can be effective in inducing remission but negative side effects preclude their long-term use. However, many times the response rates vary and disease recurrence are common and therefore there remains a significant unmet medical need in IBD. The goal for new therapies is to induce and maintain clinical remission in a greater proportion of patients than is currently achievable with standard-of-care agents. Fig. 5 summarizes some of the key small molecule compounds under development that had undergone evaluation in experimental colitis model and progressed onto successful clinical testing.
2. Signal Transduction Modulators
Signal transduction modulators are one of the major groups of targets for the development of small molecule therapy for IBD. One class that had been widely focused is the Janus kinase (JAK)/STAT pathway, which has been implicated in the etiology of many other diseases, in addition to IBD, and a number of selective compounds have entered clinical development (Fig. 5). For example, Tofacitinib is a nonselective JAK inhibitor that had been previously approved by Food and Drug Administration for rheumatoid arthritis and psoriatic arthritis. Tofacitinib potently inhibits JAK1, JAK2, and JAK3 and, to a lesser extent, TYK2 in biochemical assay (IC50=3.2, 4.1, 1.6, and 34 nM respectively). It had been shown in rat oxazolone colitis model that tofacitinib exhibit anti-inflammatory effects in vivo [67]. The effect of tofacitinib had also been shown to exhibit similar efficacy, when dosed orally or intra-cecally, in oxazolone-induced colitis in mice, but noting that intra-cecal provides colonic target engagement which do not impact splenic natural killer (NK) cell counts [68]. Consistent with the notion that oxazolone-induced colitis represents features of human UC condition, tofacitinib had been found effective for UC patients and was approved recently in May 2019 for use in moderately to severely active UC in clinical settings. Conversely, tofacitinib did not show significant improvements in remission rates, in comparison with placebo, in a phase II clinical trial for CD [69]. Another major JAK inhibitor that is currently under clinical phase III trial is Filgotinib, which has a human whole blood EC50 of 629 nM displaying a 30-fold cellular selectively for JAK1 over JAK2 [70,71]. Filgotinib efficacious effect was previously evaluated in DSS-induced colitis model and showed good in vitro ADME (absorption, distribution, metabolism, excretion) properties and high oral bioavailability in animals. In addition, upadacitinib is also another potent JAK1 inhibitor that effectively inhibits production of pro-inflammatory cytokines IL-6, oncostatin M, IL-2 and IFNγ in a variety of cellular assay [72]. The compound is currently under clinical trial phase II evaluation.
3. Cell Trafficking Modulators
Cell trafficking modulators is the other major category that is being explored for the development of small molecule treatment for IBD. For example, sphingosine-1 phosphate (S1P) is a key regulator of lymphocyte migration from lymph nodes, and blocking this pathway has been considered as a potential approach to control aberrant leukocyte migration into the mucosa in IBD [73]. In this regard, ozanimod was developed as a potent agonist of S1P1 (EC50=0.16 nM) and S1P5 (EC50=4.3 nM) and binds directly to the S1P receptor. Administration of ozanimod into mouse T cell adoptive transfer model of colitis protects the animals from body weight loss and reduced inflammation [74]. In addition, ozanimod administration at a 0.6 mg/kg in TNBS-induced colitis model in rats also protected animals from weight lost, reduce the number of circulating lymphocytes by 51%, resulting in significant improvements in disease scores [75]. With such promising results, the compound progressed into both UC and CD clinical trials, in which they are currently under phase III and phase II evaluation respectively. In addition, etrasimod is another compound designed to target S1P1, S1P4, and S1P5 receptor with EC50 values of 6.1, 147, and 24.4 nM, respectively. It was shown in scid mice receiving CD4+ CD45RBhigh T cells that administration of 3 mg/kg dose of etrasimod provided significant protection towards weight loss in the colitis-induced animals. The compound then progressed onto clinical trials (currently at phase II). Moderate-to-severe UC patients were given 2 mg dose for 12 weeks, and 43% patients showed endoscopic improvements as compared to 16.3% in the placebo group [76]. The encouraging result had led to propose extension of the clinical evaluation to CD.
α4β7 integrin is expressed on circulating T cells and mediates cell trafficking throughout the body, whereas its natural ligand is the mucosal vascular addressin cell adhesion molecule 1 (MadCAM) that is expressed predominately on the mucosal endothelium of the GI tract. Binding of MadCAM to α4β7-expressing T cells would render cell capture, adhesion and extravasation, thus leading to infiltration of pro-inflammatory T cells in the inflamed gut of IBD patients. Compounds such as carotegrast (EC50=1.4 nM) had been shown to potently inhibit the binding of α4β7 expressing cells to MadCAM. Oral dosing of methyl ester prodrug of carotegrast in mice inhibited leukocytes homing to Peyer’s patches with an EC50 of 180 nM. In IL-10 deficient CD4+ T cell transfer model of colitis, administration of carotegrast reduced IL-17, IFNγ, and TNF-α levels and significantly reduced the amount of T cells migration in the lamina propria [77]. Carotegrast has now been evaluated under a phase II study for UC and current result shows clinical remission of 23.5% versus 3.9 (placebo) at dose of 960 mg [78].
Another class of compound under development for IBD therapy that aims to mediate cell trafficking is the chemokine receptor 9. These receptors are G-protein-coupled receptor superfamily cell surfaced receptors expressed on circulating lymphocytes, and its natural ligand (CCL25) is expressed on the intestinal lumen epithelial cells. Ligand-receptor engagement results in the induction of chemotaxis of lymphocytes to the intestines. Compounds such as CCX282 had been developed to break this interaction and inhibit chemotaxis of Baf-3 cells, resulting in protection against inflammation in the TNFΔARE mice model [79]. Currently, CCX282 at 250 mg had shown promising results in a phase II study for CD, but showed less efficacy in subsequent phase III trials [80].
4. Biologics
Biological therapies have an increasing share in the modern therapeutics of various diseases including IBD. Current biological treatments generally involve either (1) blockage of proinflammatory cytokines produced by effector T cells, (2) blockage of pro-inflammatory cytokines produced by antigen presenting cells (APCs), (3) inhibition of pathogenic T cell responses via administration of anti-inflammatory cytokines, (4) targeting T cell activation, proliferation, and apoptosis, (5) enhancement of the innate immune system, and (6) inhibition of leukocyte migration to mucosal sites.
In Table 1, we have summarized some of these biologics and its responses in IBD animal models. Anti-TNF-α antibody is currently the most widely applied biologics for IBD, but interestingly this form of treatment did not exhibit encouraging results in animal models prior to the first randomized controlled trials (RCT). Studies in CD45RBhigh cell transfer model of colitis prior to RCT in humans showed only partial protection by anti-TNF-α antibody treatment with no long-term immunomodulatory effects compared with IFNγ neutralization methods [81]. TNF-α neutralization in an acute DSS-induced colitis model also exhibits no effects or even aggravated inflammation and delayed recovery [82]. In addition, anti-TNF-α antibody also failed to prevent or treat the disease in IL-10 KO mice [83]. However, subsequent evaluation of effects of anti-TNF-α therapy using other experimental colitis models confirmed its efficacy, including chronic DSS-induced colitis model (Table 1). This also shows that variability of phenotypic effect exists, and thus selection of the right animal model is crucial to demonstrate effectiveness of treatment.
In the case of targeting IFNγ, the aim is to reduce this proinflammatory product of activated T cells, NK cells and macrophages that had been shown to be elevated in human CD inflamed mucosa. Fontolizumab is a mAb against IFNγ that is currently under phase II clinical testing. Previously studies and data from experimental colitis model had demonstrated protective effect using anti-IFNγ, as seen in different model such as SAMP1/YitFc and IL-10 KO murine models [35,81,84]. However, in contrast to its protective effect, the ameliorative effect of anti-IFNγ of established colitis had not been well address. IL-10 KO colitis model showed no improvements when treated with anti-IFNγ antibody. These results inferred from animal models appeared to be in sync with the fact that fontolizumab did not reach the clinical response primary efficacy end point at day 29.
Another popular target is IL-13, given its pathogenic role in affecting epithelial tight junction, apoptosis and colonic epithelial cell restitution. IL-13 is mainly produced by NK T cells and CD4+ T cells, and eliminating its effect either through IL-13 neutralizing, depletion of NK T cells, or IL-13 deficiency in CD4+ T cells all protected mice from colitis [85,86]. In contrast to its protective effect, there has been limited positive data on ameliorating established disease. Currently, anrukinzumab and tralokinumab are under phase II trials assessing pharmacokinetics and safety in UC patients, and QAX576 (i.e., an IL-13 antagonist) is also under phase II clinical evaluation.
As mentioned above, blockage of pro-inflammatory cytokines produced by APCs is another approach towards designing IBD therapies. While mechanistically, experimental colitis model, including IL-10 KO mice model, provided evidence that the initiation and maintenance of Th1- and Th17-polarized T cell responses during chronic intestinal inflammation depends on the production of IL-12 (p35/p40) and IL-23 (p19/p40) from resident APCs. However, effects of treatment in animal models targeting anti-p40 to ameliorate established colitis are lacking. Most of the animal studies aiming at blocking APC-produced pro-inflammatory cytokines focuses on IL-12 and IL-23 through either p19, p40, and STAT-4 genetic deficiency. Antibodies against p40 (the common subunit of IL-12 and IL-23) include ustekinumab that had been shown to be more effective in both remission induction and maintenance in both CD and UC than placebo. Another antibody against p40 that was evaluated recently is briakinumab. However, studies have shown that briakinumab fail to demonstrate desired efficacy in CD and thus further studies on this asset are unlikely [87]. In addition, there are also several P19 mAbs currently undergoing clinical evaluation includes; brazikumab (MEDI2070), risankizumab (BI 6555066), guselkumab, mirikizumab (LY3074828), and tildrakizumab (MK 3222) [88].
Another key cytokine central to IBD is IL-6 that is involved in enhancing survival and resistance to apoptosis of lamina propria of T cells. Neutralizing antibodies against the IL-6 receptor (IL-6R) in experimental colitis models, including TNBS, IL-10 KO had been shown to prevent disease development and also to ameliorate established colitis [89,90]. It was suggested that the mechanism was via induction of T cell apoptosis through STAT-3 signaling pathway preventing the upregulation of anti-apoptotic gene expressions such as Bcl-2 and Bcl-xl. Current biologics targeting IL-6 associated effects include PF 04236931 (antibody against IL-6), as well as tocilizumab (antibody against IL-6R).
Similar to small compounds that prevent leukocyte migration to mucosal sites by targeting αEβ7 and MAdCAM-1, several antibodies had also been developed to achieve such effect [91]. For example, etrolizumab, which acts on β7-integrin, is currently undergoing clinical trials. It was shown that etrolizumab decreased the accumulation of CD8+ and CD4+ Th9 cells in the mice intestines more strongly than vedolizumab [92]. PF-00547569, which targets MAdCAM-1, is also currently undergoing clinical evaluation.
It is projected that many therapies will become available in the coming years if supported by the results of current clinical trials. This will provide IBD patients with a wide array of options and allow physicians to choose the best therapies for each individual patient. Experimental models of colitis continue to provide a powerful tool to indicate effectiveness and safety of these novel therapies, and the ability to infer mechanisms of action.
5. Microbiota
Gut microbiota dysbiosis represents another key factor involved in the pathogenesis of IBD and is now an attractive target for therapeutic development. Many reviews had described the use of prebiotics and probiotics to ameliorate disease using experimental models to restore the normal composition of altered microbial flora [93]. A more nascent approach is the use of synbiotics, which is a combination of prebiotics and probiotics, to achieve greater treatment efficacy. In addition, FMT is another option to achieve normobiosis restoration. Recent studies on both acute and chronic experimental colitis showed that the therapeutic effects of FMT are linked to functional modulation of the mucosal immune system and of the gut microbiota composition [94,95]. Specifically, expression of pro-inflammatory genes, antimicrobial peptides, and mucins were observed to decrease in animals receiving the treatment.
While experimental models provide a good platform to explore and develop novel therapeutics targeting the microbiota, these organisms colonize not just the intestine but also many other parts of the body. They are also affected by many other factors including food, bedding, caging, and temperature. Therefore, it is important to keep in mind the proper microbiota standardization when conducting such studies. McCoy et al. [96] recently described a protocol that aims to reduce variability and improve experimental reproducibility.
FUTURE OUTLOOK FOR THE ROLES AND USE OF ANIMAL MODELS TO STUDY IBD
1. Humanized Mouse Model
While conventional approach towards leveraging the advantages of animal model to gain insights towards both the pathogenesis of IBD through chemical induction and/or genetic manipulation of intestinal inflammation, as well as using it for preclinical studies for drug discovery and development, there had been trends towards utilizing humanize mice to speed up these efforts [97]. The aim is to mimicry the human disease physiopathology, particularly through the expression of human cytokines or immune cells, and improves bench-to-bedside transition for promising therapeutics. Humanized mouse model also may provide alternative solutions to current methods of adoptive transfer of xenogeneic (human) hemato-lymphoid cells (e.g., hematopoietic stem cells or peripheral blood mononuclear cells) into healthy wild-type mice, which on many occasions results in rapid rejection of these cells and, depending upon the type of cells injected, may result in lethal xenogeneic graft versus host disease.
Advances in this regard of using humanized mice to study IBD was demonstrated by Goettel et al., [98] on how such tools allow the direct testing of therapeutics on human immune cells which are engrafted into immunodeficient mice. The use of explants or lamina propria mononuclear cells isolated directly from human IBD patient intestinal tissue enable investigators to test therapeutic hypotheses directly on human cells and/or tissue. More recently, Goettel et al. [99] from the Snapper laboratory also showed how low-dose IL-2 can ameliorates colitis in a preclinical humanized mouse model. Current commercial humanized mouse suitable for IBD studies includes the CIEA NOG mouse® that is engrafted with human CD34+ hematopoietic stem cells.
2. Artificial Intelligence
In the new era of leveraging the power of data (e.g., publications, clinical data, high throughput data, large scale genomic data etc.), artificial intelligence (AI) and machine learning (ML) technologies had advanced and matured to analyze these large datasets. This results in increasing application of AI/ML towards both the understanding of disease pathogenesis, as well as for drug discovery and development. These might render that traditional use of expensive animal models may be subsequently replaced by these alternatives in silico method to provide inference and insights on biological research with equivalent or even better outcomes.
Similarly, these AI technologies can also be used in conjunction with animal models, (e.g., pattern recognition of animals in response to perturbation), to complement and provide greater insights and accelerate research. For example, Kozlowski et al., [100] developed an entirely automated method to score DSS-induced colitis in mice by digital image analysis of pathology slides, allowing less dependence on professional pathologist and accurate and unbiased scoring.
Indeed, AI technologies could be used to accelerate IBD research from fundamental studies including analysis of realworld data, to drug discovery and development, and in the clinic. The Nutritional Immunology and Molecular Medicine Laboratory, had created synthetic patient populations of CD and UC patients with properties of actual patient cohorts, and had built personalized predictive models of drug combinations and unravel complex relationships between diet, gut microbiome, immune system, and genetic lineup to determine the comparative treatment response in silico before testing in patients. Another example of applications of AI technologies for IBD was described by Romagnoni et al., [101] where they took the ImmunoChip dataset containing 18,227 CD patients and 34,050 healthy controls enrolled and genotyped by the International Inflammatory Bowel Disease Genetics Consortium to re-analyzed via a set of ML methods (i.e., penalized logistic regression, gradient boosted trees, and artificial neural networks) and managed to detected nearly all the genetic variants previously identified by GWAS among the best predictors, plus additional predictors with lower effects. Overall, such approach may provide a more superior alternative method to traditional experimental colitis method of understanding the disease by allowing analysis of complex systems through crunching of big data.
3. Organoid Model
Increasingly sophisticated tissue organoids can model many aspects of diseases, including IBD, which may provide an alternative method to experimental animal models. Such method not only is more cost-effective, mimics the human body’s organ structure, and also contains element of precision medicine. Use of organoid might also be beneficial in countries with strong regulations against animal testing. In addition, organoid models can also help address rare forms of IBD, especially those with very early onset in early childhood, are caused by monogenetic aberrations, for example, in IL-10, IL-10R, X-linked Inhibitor of apoptosis protein (XIAP), neutrophil cytosolic factor 2 (NCF2), or tetratricopeptide repeat domain 7 (TTC7) [102].
Several groups had demonstrated to ability to establish intestinal organoid system that can be used to understand disease pathogenesis. Specifically, Shin and Kim [103] led a study in 2018 to use his “gut-inflammation-on-a-chip” technology to confirm that intestinal barrier disruption is the upstream initiator of gut inflammation and made inference for IBD. Xu et al. [104] had also shown that transcriptional signature of epithelial organoid cultures generated from UC patients and non-IBD controls had differentially gene expression associated with epithelial secretory and antimicrobial defense to support the power of organoid system to study epithelial barrier function. In a separate study, Sarvestani et al., [105] also created patient-derived primary-organoid cultures from UC and non-IBD colonic epithelium, phenotyped them histologically and used next-generation sequencing approaches to profile whole transcriptomes and epigenomes of organoids. They concluded that these primary colonic organoid cultures from UC and nonIBD patients can be established that faithfully represent diseased or normal colonic states.
However, some of the major drawbacks of using intestinal enteroid/organoid cultures are the high cost involved. Specifically, a significant amount of expensive Matrigel, which is derived from natural polymers secreted by mouse sarcoma cells, is required to propagate intestinal enteroids/organoids. A myriad of growth factors required in the culture medium also adds on to the cost involved. In addition, both Matrigel and growth factors from conditioned media contain varying amounts of xenogeneic and undefined components, along with the potential risk of pathogen/immunogen transmission and batch-to-batch variability [106].
CONCLUSIONS
Utilizing animal models have highly provided important insights into the pathogenesis of IBD although only limited percentage of clinical interventions from animal studies were subsequently developed into something that could be fit for patient use. However, major pathogenic factors as well as inflammatory modulators are unrevealed based on the genetically engineered mouse IBD models. Alteration of multiple factors including innate immunity, adaptive immunity and epithelial defense play key roles during the development of IBD. In particular, animal models that incorporate variants of human IBD susceptibility genes have become indispensable roles to study these genes in chronic intestinal inflammation mechanistically and therapeutically. It is apparent that no single model is perfect, and therefore we need to utilize ideal/appropriate animal models to define safety and efficacy of designed novel therapeutic strategies for IBD as shown in this review article.

 XML Download
XML Download