

 PDF
PDF Citation
Citation Print
Print



INTRODUCTION
Cells, the basic unit of life, contain genetic information and express specific functions and traits depending on their genetic information. How do cells perform different functions if we don’t factor in the genetic variation? With same genetic information, cells function differently depending on gene expression. Therefore, studying gene expression at not only tissue and organ level but also in cellular level is essential. While the conventional gene expression profiling techniques detect the average gene expression level of all cell types in a given sample, single cell RNA sequencing (scRNA-seq) has the ability to reveal gene expression profile at a single-cell level, thus it is possible to uncover the cellular heterogeneity within a sample. scRNA-seq has been widely utilized to characterize individual cells of the cardiovascular system, particularly in studies involving cardiac development, disease modelling, and precision medicine. In this reviewer, we focused on how the single-cell sequencing technology can be applied in heart development, clinical field and precision medicine to treat specific diseases (Figure 1).
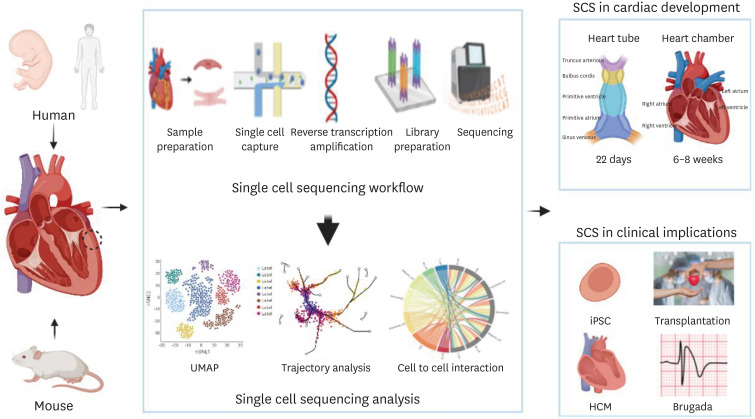
WORKFLOW OF SINGLE CELL RNA SEQUENCING
Although there is a variety of single-cell methods with distinct approaches for cell culture and amplification, the general experimental workflow of the scRNA-seq shares common steps: sample preparation, single cell capture, reverse transcription (RT) and amplification, library preparation, sequencing, and analysis.1)2) It begins with isolation of intact cells from the organ or tissue of interest, which requires a fine-tuned digest method to maximize the cell quantity and quality while minimizing cell death. Given that every tissue or cell has unique set of characteristics, the choice of proteolytic enzyme and the duration of digest must be optimized depending on cell types, culture conditions, extracellular matrix (ECM) content and cell viability.2) When cells are successfully dissociated, the number of viable cells must be >10,000 in order to confirm the data quality.2) In case of cardiomyocytes (CMs), the isolation method varies depending on age and the origin of heart tissue. The heart muscle has a larger cell size, and the human heart is more fibrous than that of mouse. Also, the frozen heart tissues are more widely utilized for single-cell transcriptional profiling compared to the fresh heart tissue due to feasible biopsy from large human cohorts.3) Next, the method of single cell capture is often determined by the cell properties. Plate-based methods include fluorescence-activated cell sorting (FACS), integrated fluidic circuit (IFC) system, ICELL8, and Manual pick-up. FACS separate individual cells into 96- or 384-well plates using scatter and fluorescence signal which enables the selection of cells of interest from dead cells.4) A major advantage of this technique is the long-term storage of cells before analysis which allows the experimental flexibility.2) Another option is the IFC system, which separates single cells into each reaction chambers. Even though the cells in the chamber are automatically lysed followed by quicker cDNA library preparation, there is a limited amount of cells that can be sorted and analyzed and only the ones less than 25 µm in diameter can be captured.4) ICELL8 (TAKARA Bio Inc., San Jose, CA, USA) system is another promising method that uses large-bore nozzle dispenser to isolate individual cells into 5,184 nano wells. It utilizes fluorescence signal imaging system to distinguish live cells from dead cells and identify wells that contain multiple cells or the ones without any cells.4)
Manual pick up selects cells with the best shape for sequencing using microscope. Although there is a minimum cell damage, some training is required for manual sorting and the throughput is quite low compared to other methods.4) As a result, the plate-based methods generally have high sensitivity and can detect up to 10,000 genes per cell. However, it has the limited number of cells that can be sorted and analyzed, and the RT must take place in individual wells which could possibly slow down the overall workflow.2)
Droplet-based methods involve the analysis of individual cells encapsulated in small oil droplets with the barcodes to each cell’s RNAs and reagents required for RT. This approach utilizes microfluidic device to perform droplet separation hence it can detect around 500–1,500 genes and the size of cells that can be analyzed is limited to 30 µm.4) Although there is a high tendency to generate doublets, droplet-based platform is more advantageous than the plate-based system in terms of throughput as it is time- and cost-effective. In case of small cells like embryonic and neonatal CMs or non-CMs (fibroblast, endothelial cells, macrophages, nuclei extracted from adult CMs), both plate- and droplet-based platforms are suitable. For larger adult CMs (120–200 µm in long axis), Manual pick-up, ICELL8, and FACS with a larger nozzle size are the most frequent used plate-based methods.4) When obtaining single cell information from adult human heart tissue, single-nucleus RNA sequencing (snRNA-seq) is also widely used due to compatibility with frozen samples.
Once the prepared single cells are captured using various methods, RT of single-cell RNA is performed to synthesize first-strand cDNA, followed by second-strand synthesis and polymerase chain reaction amplification. At the early step of RT, a short cell barcode is integrated into cDNA in order to track the cells after amplification. When using plate-based platform, each well contains well-specific barcoded RT primers while the encapsulated droplets are coated with barcoded RT primers in the droplet-based platform.5) The molecular barcoding approach involves labelling cDNA with randomly synthesized oligonucleotide (unique molecular identifier [UMI]) during RT. By counting the number of different UMIs, it is possible to identify the number of cDNAs of a gene pre-amplification, reducing amplification bias.5) These 2 barcoding strategies have mitigated the major technical limitations with early protocol; low sensitivity and low-quality cell throughput. In parallel with these technological advances, the early stages of scRNA-seq could be done more efficiently. Then, the computational workflow for analyzing scRNA-seq data involves 4 major steps, which begins with data generation, followed by data processing, exploratory analysis, and heterogeneity analysis.5)
APPLICATIONS OF SINGLE CELL RNA SEQUENCING
Detection of rare cell populations
scRNA-seq allows not only the detection of rare cells but identification of their characteristics by sequencing the gene expression level of each and every cell. While the conventional bulk RNA sequencing relies on the average data of gene expression profile of the entire cell population, single cell sequencing can provide cell-specific genetic information, uncovering the expression of genes specific to a cell type. Therefore, such large-scale single cell transcriptomics can potentially lead to the discovery of rare cell types. Hullin and his colleagues revealed distinct subsets of melanocytes, immune cells, and endothelial cells during the postnatal development of heart valve in mouse. They also found that interstitial cell populations were different in postnatal day 7 and 30, indicating that there was a significant change in gene expression and cellular functions in primordial and mature vales throughout development.6)
Identification of heterogeneity within a cell type
Even cells of the same type can exhibit different gene expression profile depending on its state or surrounding environment. In that sense, scRNA-seq technique is useful to uncover the cell-to-cell heterogeneity. In 2018, Gladka and his colleagues7) confirmed that only one subpopulation of CMs expresses myozenin 2 (Myoz2), a protein known for its role in inhibiting calcineurin, thereby restricting the hypertrophic response of CMs.8) This raised the possibility that Myoz2-enriched CMs in the epicardial surface of heart could be more resistant to calcineurin-mediated hypertrophy. Through this information, it was possible to confirm the differential gene expression profile within a cell type depending on its region and function.
Vidal Ramon from the Max Delbrück Center (Berlin, Germany) studied transcriptional heterogeneity in aging heart by comparing the single-cell transcriptomes of hearts of 12- and 18-month-old mice. Among all cell types, fibroblast exhibited the most significant difference in expression of genes involved in inflammation, ECM organization, and angiogenesis.9) Through scRNA-seq, it was able to discover the differential gene expression and underlying molecular mechanism in cells within a single type depending on its location, age, function, and disease context.10) These information function as a foundation for the development of precision medicine for treating a variety of diseases.
Understanding the cell-cell interactions
There is certainly a limitation to understand the whole phenomenon of life just by understanding the function of each cell types. In order to understand the biological phenomenon of multicellular organisms, it is vital to understand the cell-cell interactions. Unlike bulk RNA seq, scRNA-seq can be useful in figuring out the interaction between different cell types or tissues. In multicellular organisms, cells communicate via receptor-ligand interaction and the binding of ligands to their receptors activate specific cell signaling pathways which then regulate the cellular structure and function. In case of cardiovascular studies, the known cellular communication data could be useful in understanding of heart homeostasis and diseases. Skelly and his colleagues utilized lists that have been well known receptor and ligand repertoires of human cell types11) as a reference to generate lists of ligand-receptor pairs in mouse and compared it with the transcriptional data of non-myocyte cells in the adult mouse heart. As a result, he characterized numerous intercellular communication networks and identified cardiac fibroblasts as the most trophic cell population which aid the survival of specific cardiac cell population using multicellular connections.2)12) Similarly, Nona Farbehi from the Garvan Institute of Medical Research (Darlinghurst, Australia) sequenced the non-CMs fraction from murine hearts at days 3 and 7 post-sham or myocardial infarction surgery after analyzing the mouse specific cell-cell communication networks. This study confirms the non-linear dynamics in myeloid and fibroblast lineages after cardiac injury and provides further insight into cardiac inflammation, fibrosis, and regeneration.13) Although the cell-cell interaction studies using single cell sequencing neither consider the anatomical position nor provide the exact evidence of cellular crosstalk, it is still helpful in identifying multicellular signaling pathways in large-scale.2)
Single-cell trajectory inference
Cells go through temporal changes in gene expression during dynamic processes, including differentiation, disease progression, and cell cycle. Trajectory inference analyzes transcriptional changes in a single cell as a snapshot of a continuous process and places each cell in pseudo-time, an estimated temporal trajectory based on a cellular activity or transition.4) Trajectory analysis begins with dimensionality reduction, where each dimension represents a single gene expression.5) After selecting genes relevant to the cellular process of interest, the multidimensional sequencing result is transformed into a 2- or 3-dimensinal feature space using dimensionality reduction algorithms like Principal Component Analysis, T-distributed Stochastic Neighbour Embedding, or Uniform Manifold Approximation and Projection.5) Next, cells are clustered in a k-nearest neighbor graph (k-NNG) where each cell is represented as a node and each node is linked with k-nearest neighbors. Lastly, a backbone of trajectories is modelled using a graph or a curve and the pseudo-time of cells is measured by projecting cells onto the backbone.5) For example, Ren et al.14) illustrated the interaction between the major cardiac cell types and microenvironment during pathological cardiac hypertrophy and recapitulated the full spectrum of the disease progression by single-cell reconstruction of progression trajectory.
LIMITATIONS OF SINGLE CELL RNA SEQUENCING
Since its first discovery in 2009, scRNA-seq has evolved to allow more precise analysis of more cells along with the adoption of a number of tools for each step in a workflow. However, there still exists some limitations. For instance, it cannot detect the low-abundance transcripts that only 10% of the transcript is detected from a single cell and 60% of the RNA content is lost.15)16)17) In that sense, it is even harder to detect when there is a low transcript expression level. Furthermore, there is a limitation to analyze the sequencing results. As there is no absolute standard for defining the cluster of unsupervised cell populations, mathematical methods are often utilized to define the type and state of each cell and to cluster unsupervised cell population among cells in various types and function states. Although there are several clustering strategies based on machine learning algorithms, no single approach is considered as the gold standard due to unique trajectory shape and complexity in each cell population.2)
SINGLE CELL RNA SEQUENCING IN HEART DEVELOPMENT
The heart is the first functional organ to develop in a human embryo. As mesoderm cells develop into cardiac mesoderm cells and 2 endocardial tubes join together, a primitive heart tube is formed at the 21 days of gestation. It then differentiated into the early forms of hearts; truncus arterious, bulbus cordis, primitive ventricle, primitive atrium, sinus venosus, and develops into a mature heart with 4 chambers through elongation and looping processes in week 6–8 of gestation.18) In a cellular perspective, as the cardiac mesoderm differentiates into 2 types of multipotent progenitor cells, the function and the location of cardiac cells begin to exhibit different functions and fall into distinct regions. The first heart field progenitor cells become left ventricular CM, atrial CM, and cardiac conduction cell while the second heart field progenitor cells differentiate into right ventricular CM, outflow tract cell, atrial CM, cardiac conduction cell, vascular smooth muscle cell (VSMC), and endothelial cell.19)
With the ability to find rare cell populations, identify heterogeneity within one cell type, understand cell-cell interaction, and analyze cellular trajectory, the scRNA-seq technique helps to reveal the signal which affects the changes in heart traits and the differentiation process of progenitor cells during the cardiac development.20) It is also useful to sequence the human fetus heart tissue or small human adult heart tissues as it is capable of detecting the transcriptome of very few cells within a small sample size.19) CMs, which take up most of human heart atrial tissues and ventricular tissues, become distributed in various regions by the action of key genes during the cardiac development, where they have slightly different functions depending on each region.21) CMs exhibit slightly different characteristics depending on its location, that more specifically, the gene expression pattern varies rather it is located in left ventricle, right ventricle, left atrium, or right atrium. This tendency is also observable in the week 5 of gestation, that different transcription factors are expressed in atrial and ventricular CMs,22) suggesting that CMs in 4 chambers have already differentiated enough to acquire distinct spatial features. Moreover, this research team in Beijing also suggested that HMGA1 and PARP1 might have a role in regulating CM development in the early and later stage respectively.22) Litviňuková and her colleagues21) generated a human cardiac cell atlas using scRNA-seq data where the atrial and ventricular CMs were classified into 5 clusters each, reflecting the heterogeneity of atrial and ventricular CMs. They were able to determine the gene expression profile in each cluster and speculated that three types of CMs are differentiated from the second heart field when considering the molecular characteristics of 2 types of atrial CM cluster and 1 type of ventricular CM cluster.21)23)
Through scRNA-seq, it was also possible to discover the key genes that regulate the differentiation of cardiac progenitor cell (CPC) during cardiac development and act as fate determinants. The single-cell RNA analysis of Nkx2.5 and Isl1 lineages at different embryonic days revealed the role of chemotaxis-mediated intra-organ crosswalk in facilitating CPCs positioning and cardiogenesis.24) In particular, the low level of Nkx2.5 leads to defect in CM maturation and ventricular identity regulation.19)25)26) Also, it was confirmed that Mesp1 and Hand2 are the key genes involved in differentiation of pluripotent cell into cardiovascular cell and looping process of the primary heart tube respectively.27)28) Therefore, through these studies, it is not only possible to understand the cardiac development process in human, but to determine the cause of congenital heart conditions.
In order to study the cardiac development using scRNA-seq technique, the human fetal heart tissue from every week of development is needed and the non-disease sample is also required to construct an adult cardiac cell atlas. However, it is extremely difficult to do a biopsy to collect those samples. Hence, human induced pluripotent stem cells (hiPSCs) are being widely used in developmental research. hiPSCs are derived from fully differentiated somatic cells that are reprogrammed back into an embryonic like pluripotent state by expressing 4 Yamanaka factors; Oct4, Sox2, Kif4, C-Myc.29) Due to its fetal-like characteristics and ability to differentiate into cell types which lack an appropriate biopsy method, hiPSCs can be used as a key model system to study human cardiac development. Recently, several studies have identified a method to activate HOPX, a key development regulator of CMs, suggesting that stimulating HOPX might facilitate the maturation of hiPSC-CMs in-vitro.20)30) Furthermore, the data from scRNA-seq and bulk RNA suggested that NR2F2 and HEY2 promoted the earlier atrial-like and ventricular-like properties in hiPSC-CMs respectively.20)31) Although there is on-going research on identifying the regulators of CM maturation and factors that induce differentiation into specific CM subtypes, the result from in-vitro studies might not fully reflect the in-vivo human development. However, it will still be useful to model a normal and abnormal heart tissues for the subsequence endeavor in elucidating the pathogenesis of congenital heart diseases. Recent studies of heart development by using of single cell sequencing were summarized in Table 1.22)23)24)25)26)30)31)
Table 1
Summary of single cell sequencing in heart development
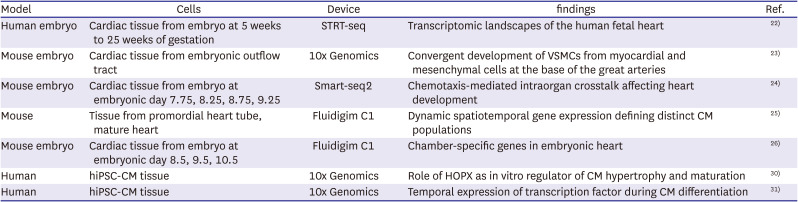
| Model | Cells | Device | findings | Ref. |
|---|---|---|---|---|
| Human embryo | Cardiac tissue from embryo at 5 weeks to 25 weeks of gestation | STRT-seq | Transcriptomic landscapes of the human fetal heart | 22) |
| Mouse embryo | Cardiac tissue from embryonic outflow tract | 10x Genomics | Convergent development of VSMCs from myocardial and mesenchymal cells at the base of the great arteries | 23) |
| Mouse embryo | Cardiac tissue from embryo at embryonic day 7.75, 8.25, 8.75, 9.25 | Smart-seq2 | Chemotaxis-mediated intraorgan crosstalk affecting heart development | 24) |
| Mouse | Tissue from promordial heart tube, mature heart | Fluidigim C1 | Dynamic spatiotemporal gene expression defining distinct CM populations | 25) |
| Mouse embryo | Cardiac tissue from embryo at embryonic day 8.5, 9.5, 10.5 | Fluidigim C1 | Chamber-specific genes in embryonic heart | 26) |
| Human | hiPSC-CM tissue | 10x Genomics | Role of HOPX as in vitro regulator of CM hypertrophy and maturation | 30) |
| Human | hiPSC-CM tissue | 10x Genomics | Temporal expression of transcription factor during CM differentiation | 31) |
SINGLE CELL SEQUENCING IN CLINICAL IMPLICATION
The advantages of single cell sequencing can be useful in clinical practice. Single cell sequencing enables drug development targeting cells that play an important role in pathophysiology, even if the cell number is small. It can also identify key genes that contribute to the disease and target them for gene therapy. As such, single cell sequencing can play a role in providing various opportunities in the field of treatment. However, there are also difficult parts in clinical application of single cell sequencing. For example, the analysis starts with obtaining living heart cells, but it is not easy to obtain human heart cells. Therefore, most of the studies analyzed through single cell sequencing in the heart field are based on animal experiments in mice. However, some studies have been conducted on human heart cells. In this review, we introduced several studies on clinical implication through single cell sequencing, along with studies on how single cell sequencing can be used in the clinical field (Table 2).14)32)33)34)35)36)
Table 2
Summary of single cell sequencing in clinical implication
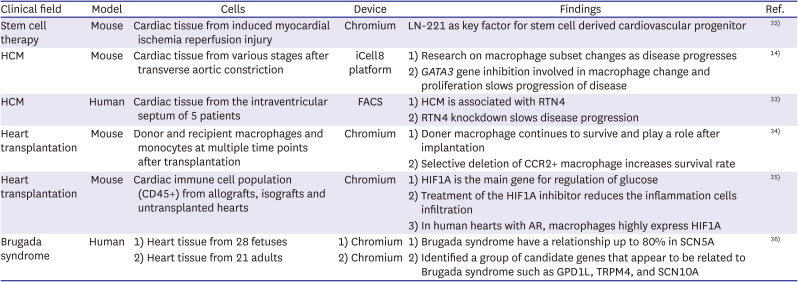
| Clinical field | Model | Cells | Device | Findings | Ref. |
|---|---|---|---|---|---|
| Stem cell therapy | Mouse | Cardiac tissue from induced myocardial ischemia reperfusion injury | Chromium | LN-221 as key factor for stem cell derived cardiovascular progenitor | 32) |
| HCM | Mouse | Cardiac tissue from various stages after transverse aortic constriction | iCell8 platform | 1) Research on macrophage subset changes as disease progresses | 14) |
| 2) GATA3 gene inhibition involved in macrophage change and proliferation slows progression of disease | |||||
| HCM | Human | Cardiac tissue from the intraventricular septum of 5 patients | FACS | 1) HCM is associated with RTN4 | 33) |
| 2) RTN4 knockdown slows disease progression | |||||
| Heart transplantation | Mouse | Donor and recipient macrophages and monocytes at multiple time points after transplantation | Chromium | 1) Doner macrophage continues to survive and play a role after implantation | 34) |
| 2) Selective deletion of CCR2+ macrophage increases survival rate | |||||
| Heart transplantation | Mouse | Cardiac immune cell population (CD45+) from allografts, isografts and untransplanted hearts | Chromium | 1) HIF1A is the main gene for regulation of glucose | 35) |
| 2) Treatment of the HIF1A inhibitor reduces the inflammation cells infiltration | |||||
| 3) In human hearts with AR, macrophages highly express HIF1A | |||||
| Brugada syndrome | Human | 1) Heart tissue from 28 fetuses | 1) Chromium | 1) Brugada syndrome have a relationship up to 80% in SCN5A | 36) |
| 2) Heart tissue from 21 adults | 2) Chromium | 2) Identified a group of candidate genes that appear to be related to Brugada syndrome such as GPD1L, TRPM4, and SCN10A |
AR = acute rejection; CCR2 = C-C Chemokine receptor 2; HCM = hypertrophic cardiomyopathy; GPD1L = glycerol-3-phosphate dehydrogenase 1 like; HIF1A = hypoxia-inducible factor 1A; LN-221 = laminin-221; RTN4 = reticulon 4; SCN10A = sodium voltage-gated channel alpha subunit 10; SCN5A = sodium voltage-gated channel alpha subunit 5; TRPM4 = transient receptor potential cation channel subfamily M member 4.
Single cell sequencing in stem cell therapy
Attempts to replace damaged cells using stem cells is currently in progress. Cardiovascular stem cell research is also underway as well. Especially, human mesenchymal stem cells have shown great opportunities in cell-based cardiac regeneration therapy due to their many advantages.37) However, heart cells have very complex interactions with surrounding cells and ECM, making it more difficult to apply stem cells than other organs. In the heart, even one cell has complex interactions with the surroundings and can have a great influence on maintaining heart function. Studies have shown that when pluripotent stem cells are transplanted into the heart, they differentiated into unintended cells or induced arrhythmia.38) Due to the characteristics of stem cells, it can be differentiated in various ways depending on the influence of the surrounding environment, so it has the possibility of differentiating into unintended cells. Therefore, single cell sequencing, which has strengths in analyzing the characteristics of each cell in a single cell unit and analyzing how it communicates with surrounding cells, is in the spotlight in the field of stem cell therapy.
A study conducted by Yap et al.32) used single cell sequencing as a tool to verify whether human stem cells differentiate into cardiovascular progenitor when given the differentiation factor laminin-221 (LN-221). They confirmed in previous studies that there was a lot of LN-221 expression in the human heart. In addition, it was found that LN-221 was highly expressed during the heart development of fetal mouse. Based on this, when LN-221 was given to human embryonic system cell lines, it was confirmed that differentiation into CM lineage cell was promoted, expression of teratoma-associated gene was reduced, and pluripotency was reduced. Since one problematic cell can affect the function of the entire heart, it is necessary to ensure that cells are differentiated into the intended cells. As single cell sequencing has an advantage in measuring the reproducibility of this single cell unit, they utilized it to check if the cells were differentiated into the intended cell types. Later, they transplanted stem cell derived cardiovascular progenitor cells into the heart of mice suffering myocardial ischemic injury. As a result, it was histologically confirmed that a human heart muscle bundle was regenerated.
Single cell sequencing in hypertrophic cardiomyopathy
Hypertrophic cardiomyopathy (HCM) is one of the major heart diseases which is mainly caused by genetic mutation. Studies have been actively conducted to find the genes involved in the development of HCM. Among the known causal genes, the β-myosin heavy chain (MYH7) and myosin binding protein C (MYBPC3) are the 2 most common and new genes related to the disease continue to be revealed.39)40) Single cell sequencing can be a suitable analysis method for finding genes involved in HCM. There are previous studies on mice for HCM. Ren et al.14) performed single cell sequencing on 11,492 cells of mice suffering from HCM. Each cell used in the analysis includes CMs and non-CMs belonging to each different stage of the disease progression. Through this study, it is proved that activation of macrophage was important for disease progression for 2 to 5 weeks, and it was confirmed that the disease progression was slowed by removing the GATA3 gene involved in macrophage proliferation from myeloid. It was known that there were various subsets of macrophage in the heart,41) but studies related to the stage of HCM were insufficient so far. A study by Ren et al.14) confirmed that inhibiting the macrophage of a specific stage can be one solution to HCM.
Wehrens et al.33) conducted a study on human HCM. They analyzed 5 human heart samples that received myomectomy through single cell sequencing. They used single cell sequencing to reveal the gene expression pattern associated with HCM. By comparing healthy cardiac cells versus cells from HCM, besides the genes that are already known to be related with HCM, they found myoglobin (encoded by MB) was associated with HCM. The hypoxic condition is probably the reason why myoglobin is expressed in HCM. Their algorithm divided CM into 6 subclusters, one of which expressed a lot of natriuretic peptide A (NPPA). Through correlation analysis, NPPA showed positive correlation with stress markers such as NPPB, ACTC1, and XIRP1. In addition, previously unknown genes, such as reticulon 4 (RTN4), were also found to have a positive correlation with HCM. RTN4 has several functions. It acts as a neural outgrowth inhibitor, causing endoplasmic reticulum stress response in human dilated cardiomyopathy (DCM) and cardiac ischemia.42) Also, it is proved that knockdown of RTN4 might be cardioprotective.43) Interestingly, positive correlation between NPPA expression and glyceraldehyde 3-phosphate dehydrogenase (GAPDH) was also observed in this study, meaning that GAPDH may not be suitable as a housekeeping gene in HCM.
Single cell sequencing in heart transplantation
Various immune cells are involved in rejection after organ transplantation. All immune cells participating in organ transplantation rejection have not yet been identified, and there is a lack of understanding of the role of these cells. Single cell sequencing can not only find a small number of cells, but also identify cell to cell communication. Therefore, in the field of organ transplantation, a small number of macrophages are found, and the role of them is well known through single cell sequencing. Until now, there are not many studies related to organ transplantation using single cell sequencing. Wu et al.44) biopsy the human kidney, which showed a transplant rejection, and analyzed it through single cell sequencing is the first paper using single cell sequencing in organ transplantation. There have been several studies on kidney and lung transplants since then, but so far, there have been a few studies on human heart transplants.45) Here, we summarized some reports. Acute rejection (AR) is an important cause of death after heart transplant. It is essential to develop an effective drug based on an understanding of immune metabolism. Kopecky et al.34) performed single cell sequencing on mouse cells showing acute cell rejection after heart transplantation. They created and analyzed barcoded count matrices using scRNA-seq (10x Genomics, Pleasanton, CA, USA). Cardiac resident macrophages are largely divided into 2 types depending on the presence or absence of C-C Chemokine receptor 2 (CCR2).46) They studied their roles, noting that donor macrophage continued to survive and play a role after transplantation. As a result, the survival rate decreased when CCR2 negative macrophage was selectively deleted but increased when CCR2 positive macrophage was selectively deleted. Based on this result, they were able to confirm that the activation of CCR2 negative macrophage among donor macrophages is beneficial for heart transplantation.
Recently, there have been studies that the results obtained through single cell sequencing in mouse hearts can be applied to human hearts. Chang et al.35) conducted scRNA-seq on CD45+ immune cells in murine isografts, allografts, and donor hearts. Through this, glucose metabolism is increased in macrophages of patients showing AR, and it is confirmed that hypoxia-inducible factor 1A (HIF1A) is the main gene for regulation of glucose. Treatment of the HIF1A inhibitor reduces the inflammation cells infiltration and the antigen presenting ability of macrophages. In human hearts with AR, macrophages of highly expressed HIF1A are also found, which can be a potential target that can effectively suppress AR.
Single cell sequencing in Brugada syndrome
Brugada syndrome is a hereditary disease characterized by ST-segment elevation in the right precordial leads and associated with life-threatening arrhythmia at risk of sudden death.47) Many pathophysiological causes are known in Brugada syndrome, but studies applied to human heart are limited. Only a single study has applied single cell sequencing to the analysis of Brugada syndrome. Tambi et al.36) conducted a single cell sequencing analysis to find the gene associated with Brugada syndrome. Their research shows that meaningful research results can be obtained using existing data sets with the analytic ability of single cell sequencing. They conducted an analysis using data sets from three different papers that previously analyzed the heart using single cell sequencing. The first data set by Cao et al.48) presents the single-cell transcriptome data derived from fetal heart tissue. This data set is consisted of four million single-cell transcriptome and only extracted 96,292 cells for the analysis of Brugada syndrome. The heart cell analysis in adults combined two single-cell sequence datasets, including adult human hearts from seven healthy potential transplantation donors and 14 deceased organ transplantation donors.21)49) By analyzing three sets of single cell sequencing analysis data, the relationship between Brugada syndrome and genes was investigated at the single cell level. As a result, 266 unique singletons and 88 repetitive gene changes were identified. It was analyzed to have a relationship up to 80% in sodium voltage-gated channel alpha subunit 5 (SCN5A). In addition, they have identified a group of candidate genes that appear to be related to brugada syndrome, such as glycerol-3-phosphate dehydrogenase 1 like (GPD1L), transient receptor potential cation channel subfamily M member 4 (TRPM4), and sodium voltage-gated channel alpha subunit 10 (SCN10A). In particular, spatiotemporal cellular heart developmental trajectory analysis shows that SCN5A, GPD1L are significantly upregulated in prenatal CMs.
PERSONALIZED MEDICINE IN CARDIOVASCULAR DISEASE
Precision medicine is an innovative approach to cardiovascular disease (CVD) diagnostics, prevention, and treatment that provides means for offering optimal care for an individual by taking one’s genetics and lifestyle into consideration. Although the term “precision medicine” and “personalized medicine” can be utilized interchangeably, it is important to note that it does not mean that therapeutics are being developed uniquely for each patient.50) Rather, the focus relies on overcoming the major issue of variable drug responses posed by traditional trial-and-error medicine. The current medical science is based on the assumption that the patients share a common patho-phenotype, thus will respond uniformly to the treatment. Yet, the extent to which a drug achieves its proper pharmacological effect largely varies among patients and most drugs prescribed in the US today are ineffective in >40% of treated patients.51) This statistic is consistent with that of CVD that at least 70% of the patients are either ineffective or partially effective to widely prescribed cardiovascular drugs, such as angiotensin-converting enzyme inhibitors and beta-blockers. Therefore, given this significant global burden, personalized medicine is expected to lead to the next generation of healthcare by 2030.52)
The rise of personalized medicine is the result of recent advances in genomic strategies. Above all, single-cell sequencing technologies in precision cardiovascular therapy have the potential to improve accuracy and efficiency by allowing single cell isolation and sensitive detection of cell-specific gene expression profiles. In human bodies, there are over trillions of cells that belong to approximately 200 different cell types.53) Individual cells are diverse with specific expression profiles that even the ones from defined cell types exhibit genomic variation. Unlike the traditional RNA sequencing techniques, which can only get the average genetic profile from all cell types, scRNA-seq has advantages of detecting heterogeneity among individual cells within a sample.
Single cell sequencing applied in precision medicine for the treatment of cancer
Early application of single cell sequencing in precision medicine primarily lies in cancer biology. It allowed for a deeper understanding of not only the fundamental genetics of cancer but also the heterogeneity across tumors from different patients and intratumor heterogeneity which can be tightly associated with the mechanisms of tumorigenesis and metastasis. A single sample of a tumor does not represent the tumor as a whole. High degree of heterogeneity among spatially apart tumor samples may also be related to diverse patient to patient responses to anti-tumor treatments and those that are resistant can cause persistence and recurrence of cancer.54) Therefore, by using single cell sequencing techniques, it is possible to reveal low abundance mutations to predict response to treatment and identify the drivers of drug resistance. Drug tolerance transition has been previously modeled in metastatic human breast cancer cell lines via single cell sequencing techniques. Paclitaxel is a common first-line treatment in breast cancer as it leads to cell cycle arrest and apoptosis.55) However, some tumor cells remain drug tolerant and resume proliferation and convert back to normalcy. The single cell sequencing result has revealed that stress tolerant cells express high cell-to-cell RNA variability with an overall gene expression profile similar to that of untreated cells with relatively high levels of RNA variants involved in microtubule polymerization and stabilization, cell adhesion, and cell surface signaling.55) The ability to profile such cell specific gene expression has the potential to illustrate the heterogeneity at the genomic and transcriptomic level which may aid and improve the prediction of treatment responses.
Single cell sequencing applied in precision medicine for the treatment of cardiovascular disease
CVD is one of main cause of mortality and morbidity worldwide, contributing to over 17.6 million deaths annually and CVD cases increased by 14.5% in 2006–2016.56) Various factors such as age genetic factor and smoking are involved in CVD. Therefore, the precision medicine is more important in CVD. The current approach to eradicate CVDs is largely based on a traditional multitiered system which involves lifestyle modifications and evidence-based therapies at large.57) Although this paradigm of medical care can alleviate symptoms and inhibit disease progression, the outcome is not certain nor promising especially in chronic illnesses involving complex and multiple causes. Only a few CVDs are monogenic and patients who share a common endophenotype might have a different phenotype when assessed at a deeper, molecular level. Thus, it must be treated differently for an optimal outcome. Therefore, it is necessary to understand the totality of CVD at a molecular level and establish a precise phenotypic profiling for a given disorder.
Multi-institutional consortia have been formed and are currently working to generate human cell atlases, including the Human Cell Atlas Consortium by the Chan Zuckerberg Initiative (Redwood City, CA, USA) and the Human BioMolecular Atlas Program (HuBMAP) by the National Institutes of Health, which will enable the characterization of changes in all cell types within the human adult heart during natural aging and in response to disease. As suggested by the cell atlases generated by the single-cell sequencing technique, the heart and surrounding vasculature are composed of multiple cell types, including CMs, fibroblasts, and endothelial cells.58) Each exhibit a different gene expression profile and contribute to different drug responses. Using this information, it becomes possible to select optimal time points and doses to maximize efficacy or detect novel variants after treatments that may potentially drive drug resistance or serve as biomarkers of therapeutic success. Here, we described one example that scRNA-seq is applied in personalized treatments.
Cardiomyopathy is often characterized as a cardiac muscle disease of unknown cause.59) However, the novel single RNA sequencing technology has allowed better understanding of the genetic etiology and pathogenesis in various types of cardiomyopathy, including HCM, DCM, and arrhythmogenic cardiomyopathy. Recently, Elliott60) revealed different transcriptional profiles of 4 clinically distinct pheno-groups of patients with DCM, underlining the need for a personalized treatment approach. Furthermore, the findings, which include 10 major cell types and 71 distinct transcriptional states from 18 healthy human hearts and 61 with DCM and ACM have provided candidate therapeutic targets for future research and improved personalized treatments.61) In case of HCM, defects in sarcomere proteins that perform the pulling motion leads to impaired pumping function of the heart.62) This results in a stress response within the muscle cells and swelling of the cardiac muscle to compensate for lost function. Therefore, patients often experience shortness of breath, chest pain and arrhythmia. In May 2022, the Huybrechts Institute applied single cell sequencing techniques on heart tissue from surgery to identify novel regulatory genes and their interactions in the heart during the disease.38) These key findings are not only useful to understand the disease-driving mechanisms in various types of cardiomyopathies but can pave the way for potential customizable therapies and provide useful information for prognosis prediction and development of novel genotype specific heart failure treatments that are more effective and have fewer side effects compared to the ones already on the market.
Despite the remarkable processes and achievements over the last few decades, personalized medicine for CVD largely remains in the research settings and the disease itself still remains the leading cause of death worldwide. To further incorporate personalized medicine into clinical workflow, it will require not only the advances in technology but better strategies for overcoming limited standardization of cardiovascular real-world data, more randomized controlled trials and education to physicians and patients.
CONCLUSION
Here, we summarized the technical information and examples of single cell sequencing applied in cardiovascular research. We described the overall workflow of the scRNA-seq technique itself and meaningful and important findings in the cardiovascular development and diseases based on scRNA-seq. Lastly, we described how the single-cell sequencing technology can be utilized in clinical field and precision medicine to treat specific diseases.
 XML Download
XML Download