

 PDF
PDF Citation
Citation Print
Print



Introduction
Advances in precision medicine have maintained pace with the development of genetic profiling technologies. Diseases that were previously classified into only a few types have gradually been subdivided into specific diagnoses, particularly in oncology, leading to an era of personalized treatment. Various therapeutic alternatives to conventional chemotherapy, such as targeted therapy and immunotherapy, have been established and meticulously investigated [1,2].
Next-generation sequencing (NGS) has markedly changed the way breast cancer is identified and treated. In addition to molecular subtyping, physicians should consider numerous genetic factors to combat breast cancer. Testing germline BRCA1/2 (gBRCA1/2) plays a pivotal role in assessing the hereditary risk of breast cancer and in guiding treatment decisions, particularly with the development of poly(ADP-ribose) polymerase (PARP) inhibitors [3,4].
Although gBRCA1/2 aberration must be confirmed under specific clinical conditions, additional costs and the necessity of blood sampling present practical hurdles to this process. Attempts have been made to infer the result of germline mutations obtained from tissue NGS results, but accuracy remains a concern. In addition, although the clinical and pathological characteristics of gBRCA-related breast cancer have been well-acknowledged over several decades [5,6], the genetic aspects of these characteristics have not been fully revealed at the NGS level.
Based on these considerations, this study was conducted to assess the consistency of gBRCA1/2 aberrations between blood germline testing and tumor tissue sequencing and to characterize the genomic landscape of breast cancer according to gBRCA1/2 pathogenicity
Materials and Methods
1. Patient selection
After obtaining approval from the Korea University Anam Hospital Institutional Review Board (No. 2017AN0401), we reviewed the charts of patients who were prescribed gBRCA1/2 testing at Korea University Anam Hospital. The test has been covered by the Korean National Health Insurance since 2012 and was conducted in patients with breast cancer having a family history of breast/ovarian cancer; diagnosed at < 40 years of age; and having bilateral breast cancer, multiple primary cancers, including breast or ovarian malignancy, or male breast cancer. The results of the gBRCA1/2 test were presented as pathogenic (gBRCA1/2p), variants of uncertain significance [VUS; (gBRCA1/2v)], and wild-type (gBRCA1/2w). Patients who agreed to participate in the K-MASTER project, a Korean National Precision Medicine Cancer Treatment Clinical Trial Platform, were enrolled in the study, and genomic profiles of tumors were collected from all patients in all groups. Detailed information about the K-MASTER project is available in the Supplementary Methods.
2. Germline BRCA1/2 test
Germline DNA was extracted from peripheral blood using the QIAamp DNA Blood Mini Kit (Qiagen, Chatsworth, CA), according to the manufacturer’s instructions. Before April 2018, germline BRCA1/2 testing based on Sanger sequencing was performed as described previously [7]. In brief, direct sequencing of 73 amplicons covering all exons and flanking introns of BRCA1 and BRCA2 was performed on the ABI 3500Dx Genetic Analyser (Applied Biosystems, Foster City, CA) using a BigDye Terminator v3.1 Cycle Sequencing Kit (Applied Biosystems). After April 2018, we conducted a multigene NGS panel test that included all exons and flanked intron regions of 31 hereditary cancer susceptibility genes, viz., ATM, BARD1, BRIP1, CHEK2, NBN, PALB2, RAD50, RAD51C, BRCA1, BRCA2, PTEN, CDH1, TP53, EPCAM, MLH1, MSH2, MSH6, PMS2, MUTYH, APC, MEN1, RET, STK11, RAD51D, TSC1, TSC2, NF1, NF2, SMARCB1, LZTR1, and VHL). NGS was performed on the Illumina MiSeq platform (Illumina Inc., San Diego, CA), generating 2×150-bp paired-end reads. Sequencing reads were aligned using the hg19 reference with BWA (ver. 0.7.10) algorithm. For the post-alignment process, duplicates were removed using Picard (ver. 1.115), and indel realignment and base recalibration were performed using GenomeAnalysisTK-Lite (ver. 2.3.9). Variant calling was performed using GATK HaplotypeCaller ver. 3.8, annotation was performed using ANNOVAR, and conversion to Human Genome Variation Society nomenclature was performed with SnpEff ver. 4.3t. Germline variants were classified according to the five-tier system of the American College of Medical Genetics and Genomics guidelines [8].
3. NGS of tumor tissue
Mutation analysis of solid tumor tissues was performed using targeted NGS. DNA was extracted, purified, and quantified from formalin-fixed, paraffin-embedded breast tumor specimens according to the K-MASTER protocol. Using the K-MASTER panel, which allows the detection of variants of 409 representative genes using the HiSeq sequencing platform, we investigated the mutation profile of the collected tissues. After passing the quality control process, the pipeline demonstrated the results of single-nucleotide variants (SNVs), copy number variants (CNVs), and genomic fusion data from each sample. Detailed laboratory and bioinformatics protocols are available in the Supplementary Methods.
In this study, the average depth of targeted sequencing coverage, duplication rate, on-target rate, pass rate score, and uniformity were 699.16 (141.51–1,202.54), 29.72% (11.8%–84.32%), 94.42% (88.74%–99.14%), 97.97% (25.9%–100%), and 77% (70%–91%), respectively. In terms of quality control, 74 of 77 samples (96.1%) covered a depth of more than 300.
4. Statistical analysis
The Student’s t test was used to compare continuous and numerical values between the groups. Normally distributed data are presented as the mean and standard deviation (SD), and deviated data are presented as the median and interquartile range. p-values were calculated using the Student’s t test or Mann-Whitney U test, based on Levene’s test. The correlation between two factors was assessed using Pearson or Spearman correlation coefficients, according to data distribution. Statistical analyses were performed using IBM SPSS Statistics software ver. 26 (IBM Corp., Armonk, NY) and visualized using R ver. 4.0.0 (R Software, R Foundation for Statistical Computing, Vienna, Austria).
Results
1. Study population
Of the 321 patients who underwent the gBRCA1/2 testing, 45 patients were categorized as harboring gBRCA1/2p, 34 were categorized as harboring gBRCA1/2v, and 242 were categorized as harboring gBRCA1/2w. After obtaining informed consent from patients and ensuring quality control of the extracted DNA, 26 gBRCA1/2p breast cancer tissues, eight gBRCA1/2 VUS tissues, and 43 gBRCA1/2w tissues were analyzed (Fig. 1). The clinicopathologic characteristics are summarized in Table 1. The median age at diagnosis was 45.5 years in gBRCA1/2p, 34.5 years in gBRCA1/2v, and 39 years in gBRCA1/2w. The difference in age between gBRCA1/2p and gBRCA1/2w group was not statistically significant (p=0.238 by t test) The familial history of breast/ovarian cancer was more prominent in the gBRCA1/2p group than in the other two groups (53.8% vs. 25.0% vs. 27.9%, respectively, having 1st degree relatives, p=0.053).

Fig. 1
Schematic flow chart of the study. Among the patients who underwent gBRCA1/2 testing covered by Korean National Health Insurance, next-generation sequencing (NGS) analysis was conducted with available tumor tissue. After quality control, 26 gBRCA1/2p breast cancer tissues, 8 gBRCA1/2 variants of uncertain significance (VUS) tissues, and 43 gBRCA1/2w tissues were analyzed.
![]()
Table 1
Sample characteristics
![]()
2. Consistency between Sanger sequencing of gBRCA1/2 testing and tumor NGS
Twelve nonsynonymous SNVs (34.3%), 11 stopgain SNVs (31.4%), 10 frameshift deletions (28.6%), and two splicing variants (5.7%) were present in all patients harboring any type of gBRCA1/2 aberration (gBRCA1/2p or gBRCA1/2v). Of the 35 gBRCA1/2 variants confirmed in the blood samples of 34 patients by Sanger sequencing, 33 variants (94.3%) were also detected in tumor tissue NGS outcomes. Five BRCA1 aberrations were inconsistent due to different reference sequences (NM_00294 and NM_007300), but all of them were the same variants after conversion. The complete profiles of BRCA1/2 aberrations are presented in Table 2.
Table 2
gBRCA1/2 mutation profile and consistency with tumor NGS assay
![]()
3. Genomic landscape of breast cancer according to gBRCA1/2 pathogenicity
Fig. 2 represents the genetic profile of breast cancer tissues and genes showing more than 5% variant allele frequency are listed. Except for BRCA1/2, TP53 was the most frequently mutated gene in all three groups (38/77, 49.4%), followed by PIK3CA (18/77, 23.4%). Variants in PIK3CA and PTEN, which are part of the PIK3CA/AKT/mammalian target of rapamycin pathway, were more prevalent in the gBRCA1/2w group than in the gBRCA1/2p group (PIK3CA 15.4% vs. 25.6%, p=0.38, PTEN 7.7% vs. 18.6%, p=0.299).
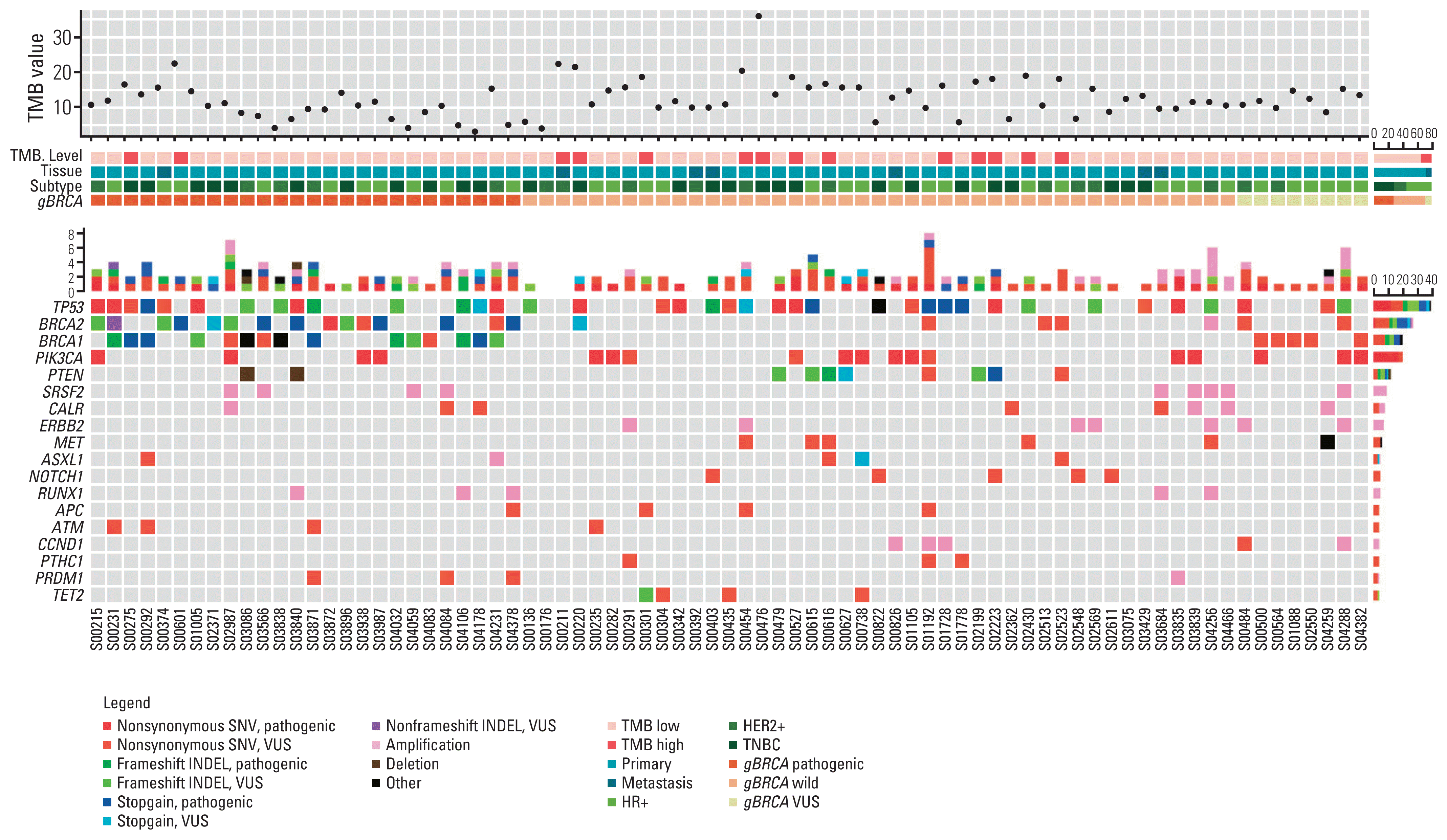
Fig. 2
Genomic landscape of genetic profile according to gBRCA1/2 pathogenicity. Heatmap visualization of genomic aberration along with tumor mutation burden (TMB) value, breast cancer subtype, and gBRCA pathogenicity. The diagram illustrates genes showing more than 5% variant allele frequency (VAF). HER2, human epidermal growth factor receptor 2; HR, hormone receptor; INDEL, insertion and deletion; SNV, single nucleotide variant; TNBC, triple-negative breast cancer; VUS, variant of unidentified significance.
![]()
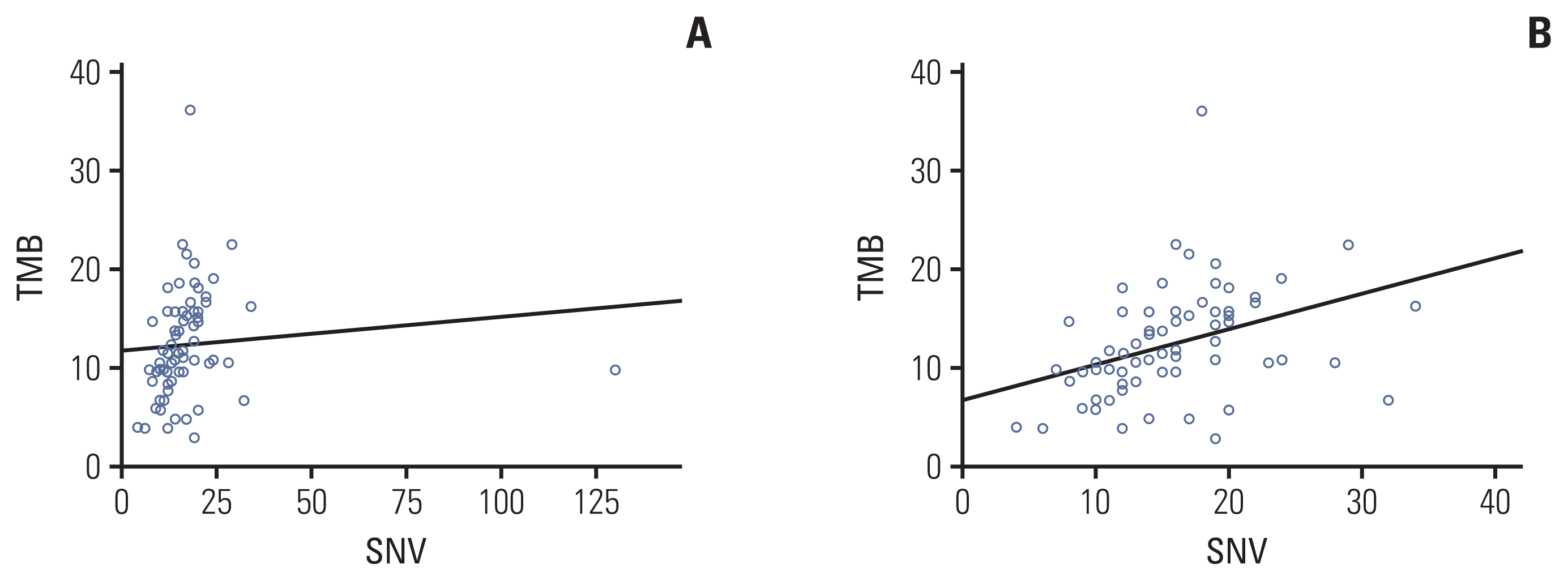
As the number of gBRCA1/2v samples was small, we compared genomic aberrancies in tumors from the gBRCA1/2p and gBRCA1/2w groups. Comparison of the two groups revealed that although the total number of reported SNVs was higher in gBRCA1/2w patients (mean, 14.81 vs. 18.86; p=0.278), the difference was not statistically significant. The total number of reported CNVs and fusions was comparable between the two groups (mean CNV, 5.77 vs. 5.47, p=0.343; fusion, 0.27 vs. 0.23, p=0.740) (S1 Table). In addition, the average estimated tumor mutation burden (TMB) was significantly higher in tumor samples from gBRCA1/2w patients than in gBRCA1/2p patients (10.21% vs. 13.47%, p=0.017). TMB value showed a linear correlation with the number of reported SNVs with marginal significance (Pearson’s correlation coefficient, 0.096; p=0.432) (Fig. 3A). However, after removing one outlier (S01192 SNV 130), the Pearson’s correlation coefficient was 0.378 and p-value was 0.001 (Fig. 3B).
4. Homologous recombination DNA damage repair gene aberration in tumor NGS according to gBRCA pathogenicity
Considering the significant differences in TMB value according to the gBRCA mutation status, homologous recombination DNA damage repair (HR-DDR) genetic variants were analyzed further. The following were defined as HR-DDR genes: ARID1A, ATM, ATRX, BARD1, BLM, BRCA1, BRCA2, BRIP1, CHEK2, FANCA, FANCD2, FANCE, FANCG, MRE11A, NBN, PALB2, RAD50, RAD51, and RAD51B. All these genes are involved in the HR-DDR pathway, as reported by Heeke et al. [9]. These genes are also included in the K-MASTER NGS panel.
Except for BRCA1/2, the gBRCA1/2p group showed only a few HR-DDR genetic aberrations, whereas tumors from gBRCA1/2w patients harbored several aberrations (Fig. 4). Among HR-DDR genes, 15 of 26 gBRCA1/2p samples (57.7%) had exclusively BRCA1/2 aberrations. Less than 50% of the patients (42.3%) harbored HR-DDR gene aberrations apart from BRCA1/2, and only four patients (15.4%) harbored multiple non-BRCA1/2 HR-DDR gene aberrations. Tumor tissues harboring multiple non-BRCA1/2 HR-DDR gene aberrations were more common in the gBRCA1/2w group (13/43, 30.2%). On average, gBRCA1/2p patients had significantly lower numbers of non-BRCA1/2 HR-DDR gene aberrations per person than gBRCA1/2w patients (0.54, 1.26, p=0.008) (S2 Table). In the gBRCA1/2w group, BRIP1 was the most frequently identified HR-related gene (23%), followed by ARID1A (16%) and BLM (12%). All HR-DDR mutation profiles are shown in S3 Table.
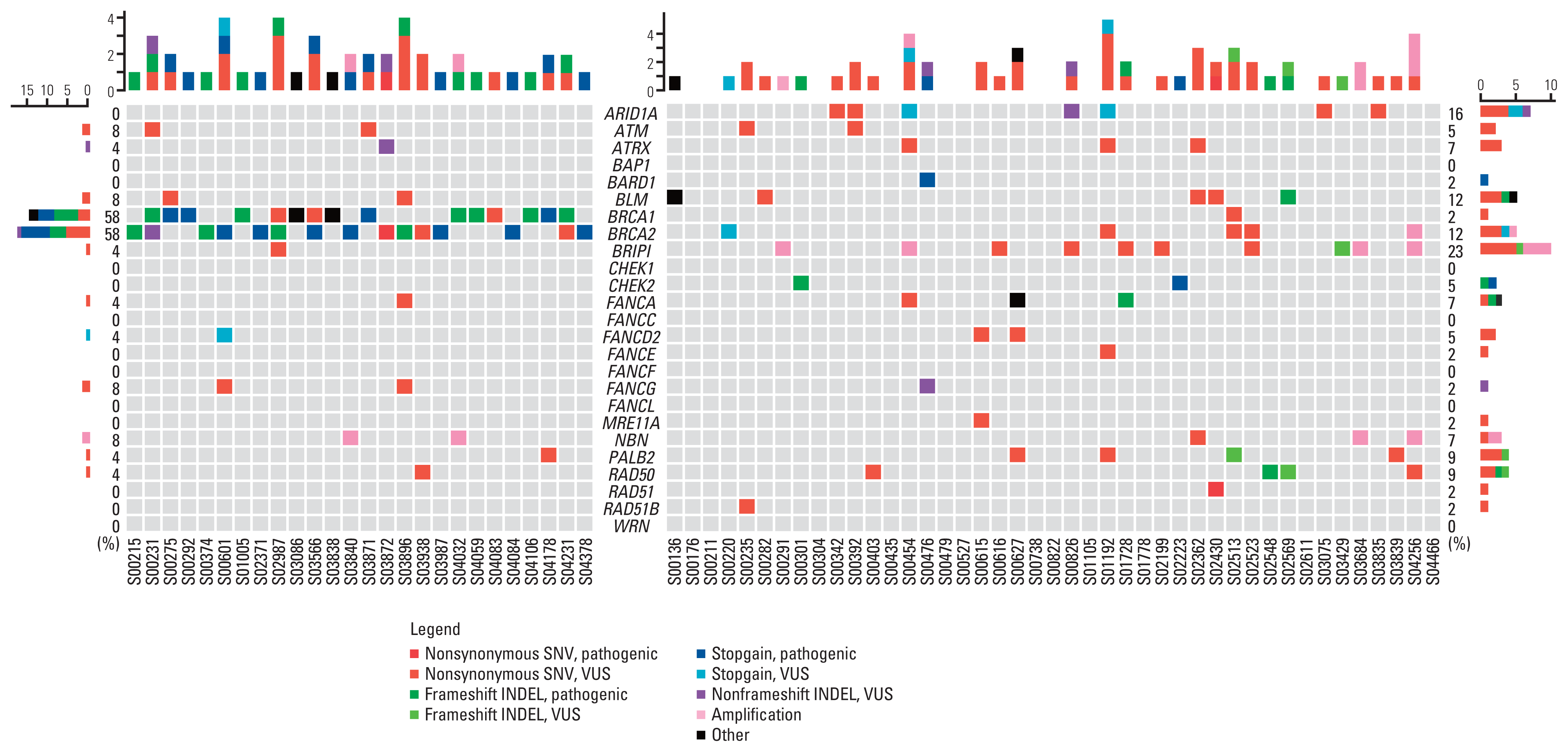
Fig. 4
Genomic landscape of homologous recombination DNA damage repair (HR-DDR) genetic profiles in gBRCA1/2p and gBRCA1/2w groups. Heatmap visualization of gBRCA1/2p and gBRCA1/2w tumor tissue confined with HR-DDR genomic aberration. INDEL, insertion and deletion; SNV, single nucleotide variant; VUS, variant of unidentified significance.
![]()
5. Correlation between TMB and homologous recombination deficiency gene aberrations
To determine whether HR-DDR gene aberrations affected DNA instability, we performed a matching analysis of TMB values based on the number of non-BRCA1/2 HR-DDR gene aberrations (S4 Fig.). Non-BRCA1/2 HR-DDR gene aberrations and TMB values showed a trend of correlation, but it was not statistically significant (Spearman’s correlation coefficient, 0.221; p=0.068). Analysis based on grouping with the number of non-BRCA1/2 HR-DDR gene aberrations is summarized in S5 and S6 Tables.
To examine the correlation between single gene mutations and TMB or SNV, we compared the average of each value between groups with or without specific genetic aberrations (S7 Table). In the analysis of TMB values with 70 tumor samples, tumor tissue with BRCA1 mutation presented lower TMB than tumors without BRCA1 mutation (p=0.016). Tumor samples with aberrations in BRIP1 showed a higher mean TMB than samples without BRIP1 mutations, but this difference was not statistically significant. None of the single gene aberrations correlated with mean SNV, possibly due to the elevated SD caused by one outlier (S01192 SNV 130).
Discussion
In this study, we analyzed breast cancer genomic landscape according to germline BRCA1/2 pathogenicity. The germline BRCA1/2 test using DNA extracted from blood samples and tumor BRCA1/2 test using tumor samples and NGS showed fair consistency (94.3%). Patients harboring the gBRCA1/2p mutation showed lower TMB values and fewer SNVs than patients with gBRCA1/2w. Although considerable differences were observed for PIK3CA and PTEN mutations, these were not statistically significant. Higher number of genes related to homologous recombination (HR) repair were mutated more frequently in the gBRCA1/2w group than in the gBRCA1/2p group, and the differences were remarkable when limited to non-BRCA1/2 HR-DDR genes. Moreover, the number of non-BRCA1/2 HR-DDR gene aberrations correlated with the number of reported SNVs.
In the current era of precision medicine, tumor NGS is frequently performed to identify potential therapeutic targets. The major goal of tissue NGS is to detect somatic mutations for actionable targets; however, information beyond somatic SNVs should also be acknowledged. Considerable efforts have been undertaken to determine germline mutations by sequencing clinical tumor samples [10]. However, discrepancies and inaccuracies in identifying germline mutations using tissue NGS have been discussed continuously. A previous study showed that tumor-only sequencing without matched normal samples could not definitively confirm germline aberrations, resulting in high false positivity [11]. Although some discrepancy is inevitable, it is proposed that the number of well-characterized pathogenic genes or their variants should be considered in tumor sequencing. BRCA1/2, MLH1, MSH2, and MSH6 are often included in cancer panels and are highly associated with inheritance of pathogenicity [12]. Our study revealed a high consistency rate (94.3%) between blood gBRCA1/2 testing and tumor tissue sequencing, suggesting that it is possible to infer germline BRCA1/2 aberration from tumor biopsy samples in clinical settings. The two inconsistent cases of our study, S01005 and S04259, presented fair quality of NGS (mean depth 794.59, 442.08 and on-target rate 90.83%, 95.72%). The inconsistency might not be due to low-quality or reference gene diversity. The only possibility is unknown human error in reporting, but clear explanation is not yet determined.
TMB is another key biomarker that can be indirectly inferred by clinical NGS. TMB is defined as the number of base alterations and indels, usually calculated by whole exome sequencing (WES). TMB calculation algorithms used in our study and their clinical efficacy were validated by comparing with TMB calculated by WES, which proved its feasibility with R2 of 0.71 [13]. Moreover, it is comprehensive with previous studies and demonstrated a fine correlation between TMB by targeted sequencing and WES (R2=0.74) [14,15]. Similarly, in our study, we assessed the matched relationship between TMB and the number of reported SNVs from a panel of tumor samples (Pearson’s correlation coefficient, 0.378; p=0.001).
TMB has emerged as a promising biomarker in the context of immunooncology, particularly in melanoma and non–small cell lung cancer [16–18]. High TMB can predict the clinical response to immune checkpoint inhibitors [19,20], but the correlation is not clear in breast cancer [21]. Approximately 20% of metastatic breast cancer cases demonstrate a high TMB [21]. In our study, 14 of 77 patients (18.2%) presented high TMB (> 16 Mb). However, the prevalence sharply increased to 27.9% (12/43) when only the gBRCA1/2 wild-type group was assessed. Taking these findings into account, further translational studies may reveal whether certain germline pathogenicity is predictive of the therapeutic effect of immunotherapy.
Hypermutated breast cancer can be driven by multiple mechanisms, including homologous recombination deficiency (HRD), and breast cancer with HRD has the highest median TMB among groups with dominant signatures [15]. In our study, the genes were selected for academic purposes and were different from those widely used commercially. With the limited resource and practical barriers with targeted sequencing, we could not clearly present HRD score. Although their important role in HR-DDR were well acknowledged in preclinical studies, genes which had not been included in approved commercial panel, such as ARID1A and NBN, should be interpreted with caution. Although BRCA1/2 are two of the essential HR-DDR genes, our data showed frequent HR-DDR aberrations and higher mean TMB in the gBRCA1/2w group than in the gBRCA1/2p group. Previous studies reported contradictory results about relationship between germline mutation and TMB. Somatic mutation of BRCA1/2 was associated with higher mutation burden and higher TMB comparing with wild type in The Cancer Genome Atlas–based analysis [22,23]. However, when the patients were classified according to germline BRCA1/2 mutation, differences of TMB was insignificant or even lower in gBRCA1/2-mutated group [24,25]. With these evidences and results of our study, we can infer that germline pathogenic variation in BRCA1/2 might contribute less to TMB than variations in HRD genes in breast cancer. As with tremendous effort to detect HRDness in solid cancer and applying the results to choosing effective treatment, the role of TMB in breast cancer should be further evaluated. The results of clinical trials evaluating the efficacy of PARP inhibitors in patients with HRDness are anticipated [26].
In our study, we noted more PIK3CA and PTEN mutations in the gBRCA1/2w group than in the gBRCA1/2m group. As two of the major cancer driver mutations, our findings provide an evidence for their mutual exclusiveness, consistent with previous studies [24,27]. A recent study also showed that breast cancer with altered phosphoinositide 3-kinase pathway harbors a significantly low rate of homologous recombination co-alterations [28]. In the future, we intend to explore the correlation between germline and somatic mutations using more genomic data from a larger sample size.
In clinical settings, many of the genes known to cause hereditary cancer syndrome are included in most cancer panels. In addition, because of convenience of its application and short turnaround time, NGS panels are increasingly being used to identify germline aberrations. Use of comprehensive genetic analysis based on WES and whole genome sequencing (WGS) is expected to increase as their accessibility has improved. These advances have expanded the possibilities of discovering novel germline mutations. Therefore, further functional genomic studies based on WES and WGS aimed at determining clinical implications of genetic variants and their effects on drug sensitivity are needed.
However, our study has several limitations. First, the sample size was too small to ensure statistical power. There had been number of patients who did their NGS analysis with blood, but they could not be included because the study was performed to investigate concordance rate of germline variants between tumor tissue and peripheral blood mononuclear cell. The statistical results should be interpreted with caution considering selection bias and relatively low prevalence of gBRCA1/2 mutation in the study cohort. Some patients were enrolled at the early stage of breast cancer, whereas others were enrolled at an advanced stage, making the total genomic profile heterogeneous. In addition, we could not verify the clinical significance of HR-DDR aberration or high TMB in terms of therapeutic targets or predictive markers. As none of the Food and Drug Administration–approved PARP inhibitors are currently reimbursed for patients with metastatic breast cancer under the Korean national health scheme, platinum-based chemotherapy remains the most affordable treatment option.
In conclusion, we herein describe the consistency between gBRCA1/2 status based on blood testing and tissue sequencing and the differences in genetic landscape according to germline BRCA1/2 gene variations. The concordance rate of gBRCA1/2 results by tissue NGS was 94.3%, and a significant difference was observed in TMB value and aberrated non-BRCA1/2 HR-DDR genes according to germline BRCA1/2 pathogenicity in patients with breast cancer. In the future, our data should be validated in a larger cohort, and the clinical impact on survival outcomes should be further elucidated.
 XML Download
XML Download