

 PDF
PDF Citation
Citation Print
Print



Introduction
Clear cell renal cell carcinoma (CCRCC) is the most common subtype of malignant tumors of the kidney, accounting for 65% to 70% of all renal cell carcinoma (RCC) [1]. The characteristic molecular alteration of CCRCC is inactivation of the VHL gene and upregulation of hypoxia-inducible factors, including vascular endothelial growth factor (VEGF) [2]. Based on the oncogenic mechanism, several tyrosine kinase inhibitors (TKIs) targeting the vascular endothelial growth factor receptor (VEGFR) signaling pathway were developed and have been widely used as the first-line treatment regimen for metastatic renal cell carcinoma (mRCC). For example, sunitinib, one of the TKI inhibitors, has demonstrated an objective response rate (ORR) of 42%, which exceeds the ORRs of conventional cytokine therapy (5% to 20%) [3]. However, despite the initial response to TKI therapy, most mRCC patients experience drug resistance within 6 to 15 months after treatment due to the development of acquired resistance to TKIs, requiring alternative therapeutic strategies [4].
Recently, immune checkpoint inhibitors, including anti–programmed death-1 (PD-1) and programmed death-ligand 1 (PD-L1) monoclonal antibodies, have shown considerable therapeutic potential in various types of malignancies, such as non-small cell and small cell lung cancers, urothelial carcinoma, and breast cancers [5]. PD-L1 is a key immune checkpoint molecule expressed on tumor cells and tumor-infiltrating immune cells. Binding of PD-L1 to its receptor PD-1 on tumor-specific T cells induces T-cell tolerance and helps tumor cells avoid immune destruction [6]. Previous clinical studies of advanced metastatic clear cell renal cell carcinoma (mCCRCC) have demonstrated a survival benefit of PD-1/PD-L1 inhibitors as monotherapy or in combination with other active agents, with ORRs ranging from 36.4% to 59.3%, as first-line treatment [7,8]. However, TKIs are still recommended as the first-line treatment for patients who cannot receive or tolerate immune checkpoint inhibitors [9].
While trying to define the TKI-resistance mechanism by comparing gene expression of paired RCC tissues harvested before and after TKI treatment, we found that PD-L1 expression was increased in a proportion of patients with progression of mCCRCC after TKI treatment. The findings encouraged us to explore the underlying mechanism of PD-L1 expression in TKI-resistant mCCRCC.
Materials and Methods
1. Patients
As a discovery cohort, we retrospectively searched for mCCRCC patients treated with VEGFR-TKIs from whom tumor tissues were harvested before and after the TKI treatment period. To define differential gene expression responsible for TKI resistance, we included only those patients from whom post-TKI mCCRCC tissue was harvested within a month after the diagnosis of progressive disease after TKI treatment. Although post-TKI biopsy is not usually performed once the diagnosis of CCRCC is confirmed at pretreatment period, we were able to find 10 patients that satisfied the above inclusion criteria among 553 patients with recurrent or metastatic CCRCC who were treated with VEGFR-TKI at the Asan Medical Centre, Seoul, Republic of Korea, from 1997 to 2013. Pre-TKI tumor samples were obtained from the primary kidney tumor in six patients and from metastatic tumors of the brain, cheek, leg, and lung in the remaining four patients. In cases in which the tumor tissues were biopsied more than once before TKI treatment, the sample obtained closest to TKI treatment was chosen as the pre-TKI tumor sample for further analyses. The post-TKI tumor samples were biopsied from metastatic tumors in the ileum in two patients; the lung in two patients; and the scalp, retroperitoneum, brain, stomach, thigh, and abdominal wall (n=1 each) in the remaining six patients.
The patients’ clinical information was obtained from electronic medical records and/or hospital charts. Each patient was reviewed according to the 2016 World Health Organization (WHO) Tumor Classification, graded according to the International Society of Urological Pathology (ISUP) grading system, and staged according to the American Joint Committee on Cancer (AJCC) Staging System, 8th edition.
2. Gene expression profiling and pathway analysis
Total RNA was extracted from the 10 pairs of pre- and post-treatment formalin fixed and paraffin embedded tumor tissues of the discovery cohort. Gene expression profiling analyses were performed using Human Genome Human Transcriptome Array 2.0 (Affymetrix, Santa Clara, CA) according to the manufacturer’s instruction at an outside laboratory under the supervision of two authors (H.G. and H.S.H.). The raw data were normalised by the robust multiarray average method implemented in Transcriptome Analysis Centre ver. 3.0 (Affymetrix). The probes were annotated using.chip files for HTA 2.0 platform for gene set enrichment analysis (GSEA) provided by the Broad Institute (http://software.broadinstitute.org/gsea/index.jsp). The differentially expressed genes (DEGs) in the PD-L1–expressing and –nonexpressing groups were investigated by an empirical Bayes moderated t-test using R package limma [10]. The cut off criteria for the DEGs were < 0.05 of normal p-value instead of the false discovery rate (FDR) because of the small number of patients. No annotated genes were excluded.
To determine the enriched pathways for PD-L1 expression, both the hypergeometric test and GSEA were carried out. Hallmark and canonical pathway gene sets (C2cp) of the Molecular Signatures Database (MSigDB) ver. 6.0 (Broad Institute, Cambridge, MA; http://www.broad.mit.edu/gsea/msigdb/index.jsp) were used for the pathway analysis. Gene set permutation rather than sample permutation was performed during the analysis because of the small number of patients. An FDR below 0.05 was considered significant enrichment.
3. Immunohistochemistry in the tissue microarray cores and the whole-tumor sections
All available specimens were formalin-fixed and paraffin-embedded and used for tissue microarray (TMA) construction, which consisted of three representative cores 1.5 mm in diameter. The tumor areas were randomly selected while trying to exclude necrotic and degenerative areas and maximize tumor cell content. To exclude the issue of intratumoral heterogeneity, immunohistochemistry was also performed on whole-tumor sections when residual tumor cells were available.
The discovery cohort was assessed using the anti–PD-L1 antibody (clone E1L3N, 1:100, rabbit monoclonal, catalog No. 13684, Cell Signaling Technology, Danvers, MA) on the TMA construct. In the whole-tumor sections, PD-L1 expression was assessed using the anti–PD-L1 antibody E1L3N and a US Food and Drug Administration–approved PD-L1 assay (22C3 pharmDx, code SK006, Agilent, Santa Clara, CA).
Immunohistochemical (IHC) staining was performed using the BenchMark XT automated staining system (Ventana Medical Systems, Tucson, AZ) according to the manufacturers’ protocol. The nuclei were counterstained with hematoxylin.
The IHC staining results were assessed in a semiquantitative manner. PD-L1 expression was first evaluated according to the intensity (negative, weak, moderate, or strong) and the percentage of positive tumor cells and immune cells, including lymphocytes and macrophages. Either tumor cells with membranous patterns or immune cells with cytoplasmic or membranous patterns showing more than weak intensity in > 1% were considered positive for PD-L1 expression [11]. The investigator was blinded to the associated clinicopathological information.
4. Cell culture
The human RCC cell lines 769-P, 786-O, ACHN, Caki-1, and A704 were purchased from the American Type Culture Collection. A704 was cultured in modified Eagle medium (Gibco, Grand Island, NY), and the others were cultured in RPMI-1640 medium (Gibco) supplemented with 10% fetal bovine serum (FBS; Gibco) and 1% penicillin/streptomycin at 37°C in a humidified atmosphere containing 5% CO2. To develop TKI-resistant RCC cell lines, a dose-response study was first performed to assess their sensitivity to sunitinib (Sutent, Pfizer Pharmaceutical, New York, NY), which is the most frequently administered TKI in mCCRCC, by treating them with various concentrations from 0.1 to 50 μM for 72 hours; then the 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl tetrazolium bromide (MTT) assay was performed to estimate the half-maximal inhibitory concentration (IC50) value. The cells were then treated with sunitinib for 6 months at various concentrations ranging from 0.5 to 5 μM, which were chosen to span the IC50 value. The culture medium was changed every 3 days. Sunitinib-sensitive parental cell lines were used as control cell lines. Sunitinib-sensitive parental and sunitinib-resistant cell lines were designated as suS and suR, respectively.
5. MTT assay
The proliferation activity of RCC cell lines was evaluated by MTT assay (Sigma Chemical Co., St. Louis, MO). Briefly, cells (2×104) at the exponential growth phase were seeded in 96-well plates and allowed to attach overnight at 37°C. MTT reagent was added to each well and incubated for 4 hours at 37°C. The supernatant was discarded; the MTT-formazan crystals formed by metabolically viable cells were solubilized with 100 μL of dimethyl sulfoxide (DMSO) on a shaker for 10 seconds; then absorbance was measured in an enzyme-linked immunoassay microreader at 540 nm. Each experiment was performed in triplicate and repeated three times for reliable comparison.
6. Mammalian target of rapamycin inhibitors and activator
A dual phosphoinositide 3-kinase (PI3K) and mammalian target of rapamycin (mTOR) inhibitor, dactolisib (NVP-BEZ235), and an mTOR inhibitor, everolimus (RAD001), dissolved in DMSO were purchased from KIM & FRIENDS (Seoul, Korea) and used at final concentrations of 0.5 μM and 5 nM, respectively based on earlier studies [12–14]. An mTOR activator, MHY1485 (synthesised 4,6-dimorpholino-N-(4-nitrophenyl)-1,3,5-triazin-2-amine; Sigma-Aldrich), dissolved in DMSO was used at a final concentration of 2 μM [15].
7. Western blotting
The following antibodies were used in western blotting: rabbit antibodies against PD-L1 (1:100 dilution), phospho-mTOR (Ser2448) (p-mTOR; 1:100 dilution) and phospho-S6 ribosomal protein (Ser 235/236) (p-S6RP; 1:100 dilution) from Cell Signal Technology (Beverly, MA). Whole cell extracts were prepared using radioimmunoprecipitation assay lysis buffer (89900, Thermo Scientific, Waltham, MA) and protease inhibitor cocktail (BPI-9200, Tech & Innovation Co., Chuncheon, Korea). Total protein was subjected to electrophoresis on 12% (PD-L1 and p-S6RP) and 8% (p-mTOR) of sodium dodecyl sulphate–polyacrylamide gel electrophoresis gels for 2 hours at 80 V. β-Actin served as the internal control.
8. Clonogenic survival assay
The cells were plated onto six-well plates in triplicate at low density (0.5×103 cells per well). After 2 weeks in culture, the colonies were stained with 1.0% crystal violet for 15 minutes, then the grossly visible colonies were counted manually. The clonogenic survival fraction (SF) was calculated as follows: SF=(number of colonies formed at a given dose/number of cells plated at a given dose)×(control number of cells plated/control number of colonies formed) [16].
9. Wound healing assay
The cells were plated onto six-well plates at 8×105 cells in triplicate. After overspreading on the plate, the cells were wounded by drawing vertical scratches with a plastic pipette tip, then incubated in regular medium. Migration of cells was observed and photographed under an optical microscope regularly.
10. Matrigel invasion assay
A total of 100 μL of serum-free medium including Matrigel was added to each well of a 24-well plate containing inserts with 8-μm pores and incubated overnight at 37°C to form an artificial basement membrane. Then 2.5×105 cells were seeded in triplicate on the upper chamber of the transwell, and 10% FBS-containing medium was added to the lower chamber. Following 48 hours of incubation at 37°C, the cells remaining on the top of the inserts in the upper membrane were removed by wiping with a cotton swab. Those cells invading the underside of the membrane were fixed in 3.7% formaldehyde, then stained with 1% crystal violet. Cells penetrating the Matrigel were photographed.
Results
1. Characteristics of patients in the discovery cohort
The clinicopathological features of the 10 patients in the discovery cohort are summarised in Table 1. The median age at initial diagnosis was 51.8 years (range, 40 to 66 years), with a 4:1 male/female ratio. At initial diagnosis, three patients presented with metastatic disease, but all patients eventually developed distant metastases (median, 2 months; range, 0 to 169 months). Four patients had received treatment prior to TKI therapy, including treatment with interferon (two patients), interleukin-2 (one patient), and radiation (one patient). As the TKI regimen, seven patients received sunitinib and three patients received pazopanib.
The pre-TKI tumors were of high grade (WHO/ISUP grades 3 or 4) in six patients, with accompanying sarcomatoid features observed in three patients. Compared with pre-TKI tumors, post-TKI tumors demonstrated aggressive pathological features, with more patients with high-grade tumors (nine patients) and tumors with sarcomatoid features (six patients).
The patients developed tumor progression at a median of 13.5 months after initiation of TKI (range, 1 to 70 months), and all died of the disease at a median of 24.5 months after progression (range, 5 to 96 months).
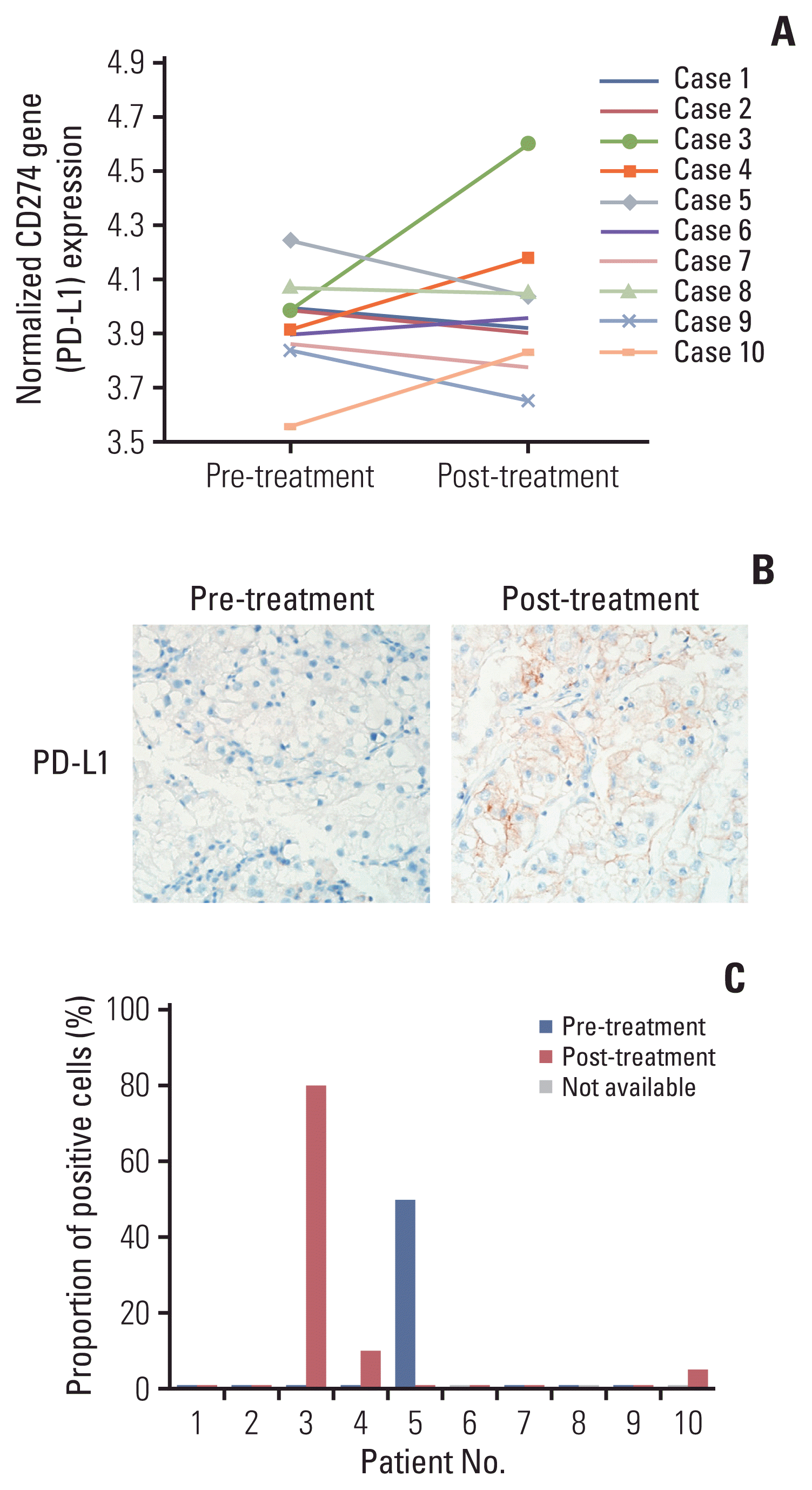
2. Expression of PD-L1 was increased in a subset of TKI-resistant CCRCC patients in the discovery cohort
Global gene expression was compared by microarray experiments on paired tumor tissues harvested at pre-TKI treatment and those harvested at progression of the disease after TKI treatment. Among DEGs, increase of PD-L1 expression in post-treatment tissues was identified in three patients (patients 3, 4, and 10) (Fig. 1A). To verify the microarray results at the protein level, PD-L1 IHC staining was performed on the 10 paired patients. In TMA, PD-L1 expression of tumor cells was noted in the post-treatment tissues of three patients. Patient 3’s tissue yielded a membranous pattern and the other patient samples yielded mixtures of cytoplasmic and membranous patterns (Fig. 1B and C). In consideration of tumor heterogeneity, the PD-L1 IHC staining was performed using whole-tumor sections. In addition to the three patients who were positive in terms of TMA, patient 6 was confirmed to be positive in terms of tumor cells (S1 Table). Therefore, an increase in PD-L1 expression in post-treatment tissues was noted in four patients. Both anti–PD-L1 antibodies, E1L3N and 22C3, showed similar expression patterns in the whole-tumor sections.
PD-L1 expression of immune cells was noted in the post-treatment tissues of two patients. In the TMA construct, PD-L1 expression was noted in only one patient (patient 5) in both pretreatment and post-treatment tissues. Another patient (patient 6) showed PD-L1 expression in the immune cells of the whole-tumor section. However, PD-L1 expression status could not be assessed in the pretreatment needle biopsy tissue because the tumor tissue was not available.
3. DEGs associated with PD-L1 upregulation
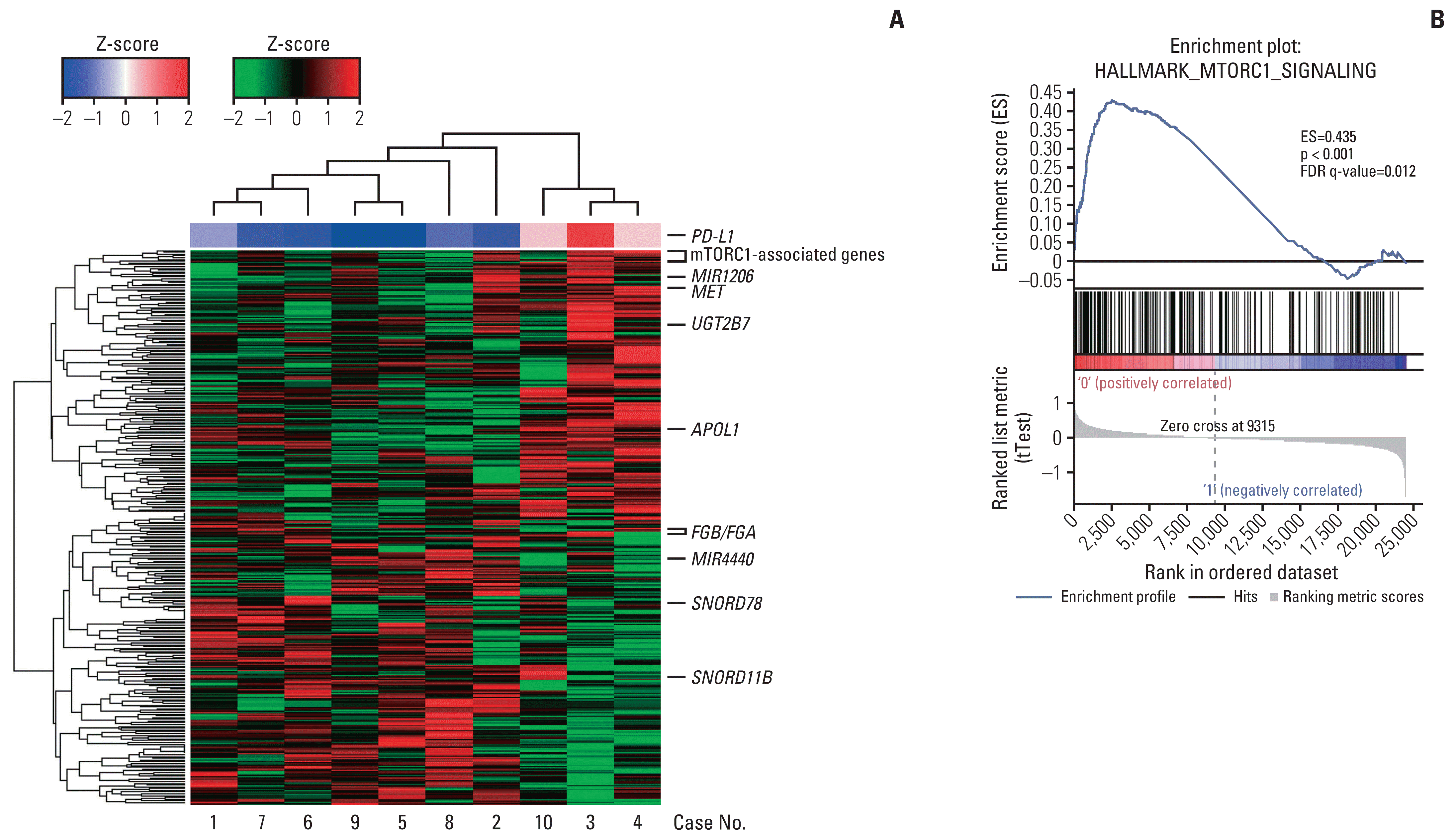
To define DEGs associated with PD-L1 upregulation, the global gene expression of post-treatment CCRCC tissues was compared between three patients whose PD-L1 increased and the remaining seven patients whose PD-L1 expression was not increased in the TKI-resistant post-treatment tissues. When the selection criterion of nominal p-value < 0.05 was applied, 434 DEGs were obtained (Fig. 2A, S2 Table).
The analysis of DEGs in the discovery cohort by the hypergeometric test and GSEA resulted in the selection of mTOR complex 1 (mTORC1) signaling, integrin signaling, hypoxia and glycolysis as the gene sets positively associated with PD-L1 upregulation (Table 2, S3–S5 Tables). Since mTOR inhibitors have been widely used to treat patients with advanced RCC, the mTORC1 signaling pathway was chosen for further study (Fig. 2B). Among the DEGs, those overlapping with the hallmark mTORC1 gene sets on GSEA were GBE1, PLOD2, HSPA9, PDAP1, HSPA5, NMT1, PSMC2, TES, ACLY, and HSPD1. The gene sets negatively associated with PD-L1 upregulation included those associated with allograft rejection, graft-versus-host disease, cytotoxic T lymphocyte antigen-4, T-cell receptor and various interleukin signals, which suggests a decreased tumor immune response (S6 Table).
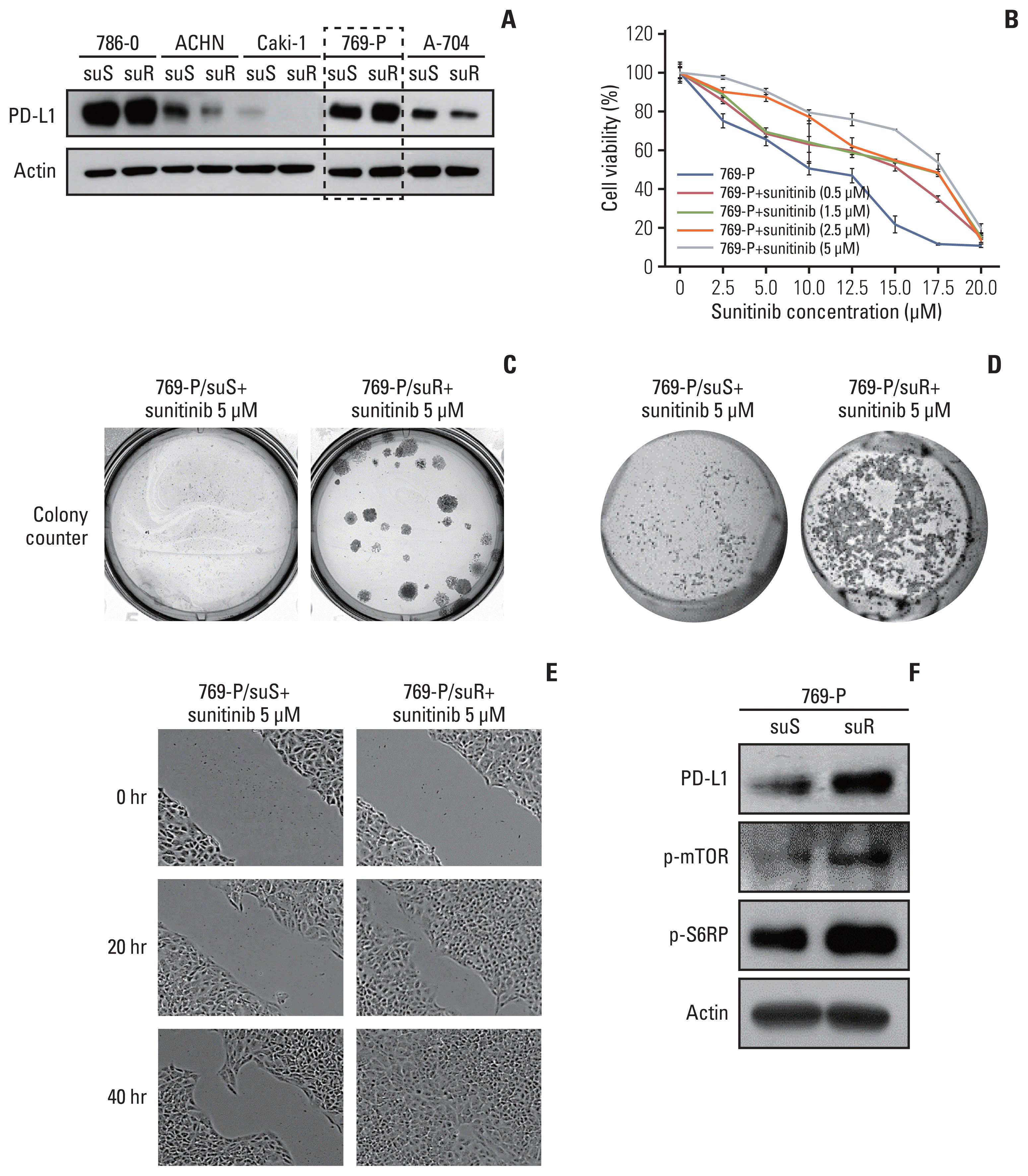
4. Development of VEGFR-TKI–resistant CCRCC cell line with PD-L1 upregulation
To examine the effect of TKI resistance on the tumoral behaviour of CCRCC, TKI-resistant CCRCC cell lines were developed by long-term treatment of five RCC cell lines, 769-P, 786-O, A-704, ACHN and Caki-1, with sunitinib. Although sunitinib resistance was successfully established in these five cell lines, upregulation of PD-L1 was noted only in 769-P in their corresponding sunitinib-resistant cells (Fig. 3A). After long-term treatment with 0.5, 1.5, 2.5, and 5 μM sunitinib, the IC50 of 769-P sublines increased to 15, 15.4, 15.5, and 17.7 μM, respectively, compared with 10 μM for 769-P/suS (Fig. 3B). Therefore, 769-P cells treated with 5 μM sunitinib were chosen for further experiments.
5. Sunitinib-resistant 769-P with aggressive behavior and upregulation of mTOR signaling
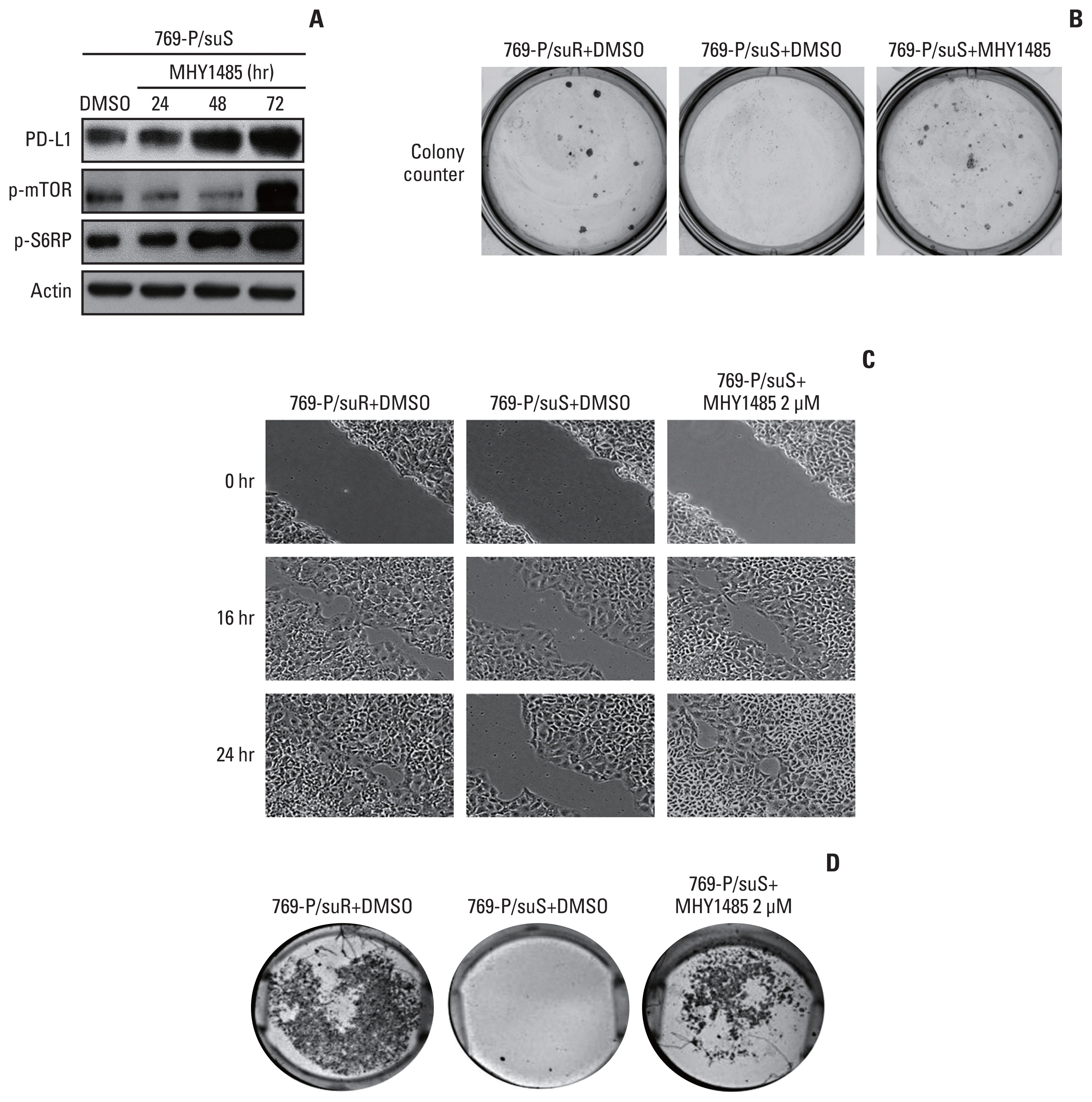
In addition to its high proliferative capacity (Fig. 3B), 769-P/suR showed higher clonogenic survival with greater numbers and larger sizes of colonies in the colony-forming assay compared with 769-P/suS (Fig. 3C). Furthermore, 769-P/suR showed more cells invading the Matrigel membrane in the Matrigel invasion assay and migrating rapidly into the scratched area in the scratch test assay, suggesting its higher invasion and migration capacities (Fig. 3D and E). In addition to PD-L1 upregulation, the expression of p-mTOR and p-S6RP, which has been used as a surrogate marker for mTORC1 activity [17], was also increased in 769-P/suR (Fig. 3F).
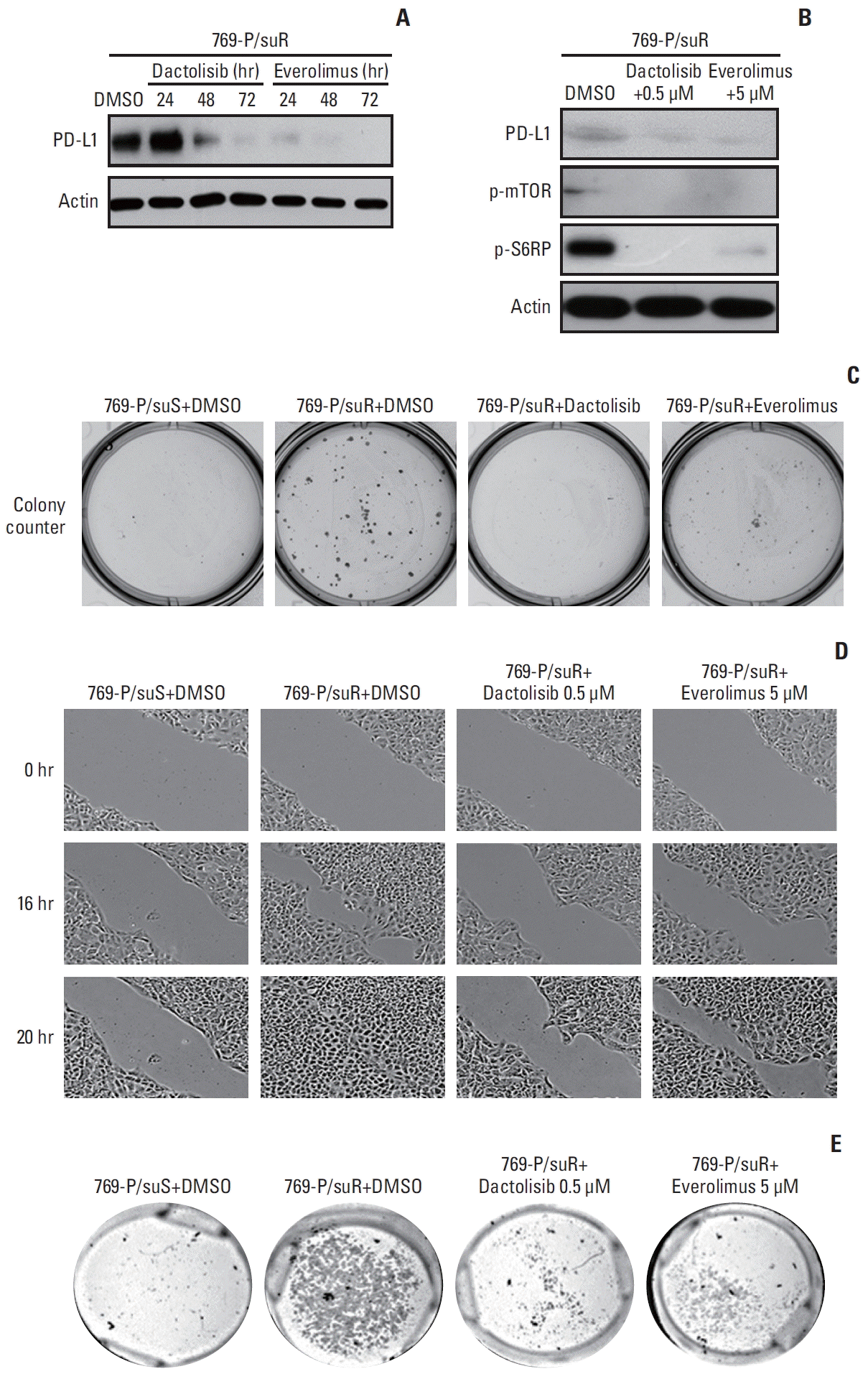
6. Inhibition of the mTOR pathway down-regulates PD-L1 and suppresses aggressive behavior of VEGFR-TKI–resistant CCRCC
To evaluate whether PD-L1 expression was dependent on activation of the mTOR signaling pathway, 769-P/suR was treated with two pharmacologic inhibitors, dactolisib and everolimus. They inhibited PD-L1 expression in a time-dependent manner (Fig. 4A). In addition, they inhibited the expression of p-mTOR and p-S6RP (Fig. 4B). Treatment of 769-P/suR with dactolisib and everolimus resulted in smaller numbers and sizes of colonies in the colony-forming assay (Fig. 4C) and inhibited migration and invasion in the scratch test assay and the Matrigel invasion assay, respectively (Fig. 4D and E).
7. Stimulation of the mTOR pathway up-regulates PD-L1 expression and represses aggressive behavior of VEGFR-TKI–resistant CCRCC
Stimulation of the mTOR pathway with MHY1485 increased the expression not only of p-mTOR and p-S6RP, but also of PD-L1, in a time-dependent manner in 769-P/suS cells (Fig. 5A). MHY1485 stimulation increased the number and size of colonies in the colony-forming assay (Fig. 5B) and promoted migration and invasion in the scratch test assay and the Matrigel invasion assay, respectively (Fig. 5C and D).
Discussion
We demonstrated that PD-L1 expression was increased in a subset of TKI-resistant CCRCC patients. An in vitro study using a TKI-resistant CCRCC cell line revealed that mTOR signaling increased PD-L1 expression and was responsible for aggressive tumoral behavior. These results may provide an explanation for the clinical therapeutic effects of the use of combination therapy with TKIs and mTOR inhibitors to prevent PD-L1 expression and the aggressiveness of TKI-resistant CCRCC.
VEGF has been isolated as an endothelial cell-specific mitogen that mainly functions through binding to VEGF receptors on endothelial cells [18]. The therapeutic effect of VEGF-targeted therapy has been largely explained by its antiangiogenic effect [19]. In addition, VEGF-mediated autocrine signaling has been reported in breast carcinoma, squamous carcinoma, and glioma to promote the growth, survival, migration, and invasion of tumor cells [20–22]. The previous reports are in accordance with our results, in that an endothelium-independent mechanism explains, at least in part, resistance to VEGFR-TKI in CCRCC.
VEGF is also known to play an important role in immune modulation and to establish immune privilege of tumor cells by inhibiting the infiltration of T cells and blocking dendritic cell differentiation [19]. Paradoxically, VEGFR-TKI treatment resulted in upregulation of PD-L1 expression in a subset of CCRCC patients and RCC cell lines in this present study. In a previous study, RCC patients treated with antiangiogenic therapy showed enhanced expression of PD-L1 compared with untreated patients, which was inversely correlated with patient survival [23]. These findings suggest that TKI-resistant CCRCC develops an immune evasion mechanism by upregulation of PD-L1 expression through the mTOR signaling pathway.
As a downstream effector of the PI3K/AKT pathway, mTOR exerts its function via protein synthesis and affects cell proliferation, angiogenesis, and metabolism [24]. It acts through two distinct multiprotein signaling complexes, mTORC1 and mTOR complex 2. mTORC1 activates ribosomal S6 kinase (p70S6K) and inhibits a translational repressor eukaryotic translation initiation factor 4E-binding protein 1 (4E-BP1), which eventually initiates translation. Through its effect on protein synthesis, mTORC1 modulates the activity of cell-cycle–regulating proteins, including hypoxia-inducible factor 1α and VEGF [25]. Our results imply that the increased mTOR signaling pathway may serve as an alternative oncogenic mechanism in a subset of TKI-resistant CCRCC patients in spite of the inhibition of the major oncogenic VEGF signaling pathway by VEGFR-TKI. They also suggest that combination therapy with VEGFR-TKIs and mTOR inhibitors might help prevent PD-L1 expression and the aggressiveness of TKI-resistant CCRCC.
Upregulation of PD-L1 through the mTOR pathway has been reported in non–small cell lung cancer. In an IHC analysis of human lung cancer where p-S6RP expression was used as a marker of active mTOR signaling, 40% of lung cancer patients had active mTOR signaling and PD-L1 expression, with a statistically significant correlation between the two markers. mTOR activation and PD-L1 expression were detected in non–small cell lung cancer cell lines, where PD-L1 expression was not only decreased by inhibition of the PI3K/Akt/mTOR pathway but was also induced by epidermal growth factor and interferon γ in an mTOR-dependent manner [26].
Clinical studies on concomitant treatment with VEGFR-TKI and mTOR inhibitors have been conducted. Phase I trials of sunitinib with everolimus or temsirolimus resulted in significant acute and chronic dose-limiting toxicity, which prevented their clinical adoption. A recent phase II study of combined therapy with another TKI, lenvatinib, plus everolimus found a progression-free survival benefit, with an ORR of 43%, and the combination therapy received Food and Drug Administration approval for the treatment of advanced RCC after failure of prior TKI therapy [27].
In our study, only a subset of TKI-resistant CCRCC patients showed upregulation of PD-L1 expression in the tumor cells at the mRNA level (3 of 10 patients) and at the protein level (4 of 10 patients). Although PD-L1 expression of immune cells in post-treatment tissues was noted in two patients, neither of the patients were confirmed to have increased PD-L1 expression in the immune cells of post-treatment tissues compared with pretreatment tissues. Previous studies have reported PD-L1 expression of immune cells to be associated with worse prognosis, but the significance of such expression is uncertain in the context of TKI-resistant CCRCC [28].
In accordance with our results, clinical studies showed that PD-L1 inhibitors were effective in a proportion of patients with metastatic CCRCC who developed drug resistance after treatment with VEGFR-TKI inhibitors. The ORRs of the PD-L1 inhibitors (atezolizumab and avelumab) were 36% to 55.2% [8,29]. Nivolumab was the first PD-1 inhibitor, and its ORR was 41% to 42% in patients with advanced RCC [8,29]. As shown in the present study, the authors reported that RCC with increased PD-L1 expression was associated with worse prognoses [30,31], although drug efficacy was observed irrespective of PD-L1 expression [32].
The present study had some limitations. Although we demonstrated upregulation of PD-L1 and mTOR signaling in a subset of TKI-resistant CCRCC patients using post-treated CCRCC tissue compared with matched pretreated tissues, the number of these patients was small (10 patients only). It is mainly because once the diagnosis of CCRCC is confirmed at pretreatment, responsiveness to treatment is determined clinically by imaging studies without pathologic examination. Despite the limitation, this study provides an insight into the acquired resistance mechanism of mCCRCC. Therefore, prospective multi-institutional studies need to be performed to collect a large number of paired samples to confirm the results of the present study. In addition, future prospective studies will allow procurement of viable tumor cells and in vivo studies using animal models.
 XML Download
XML Download