

 PDF
PDF Citation
Citation Print
Print



Introduction
Materials and Methods
1. Patients
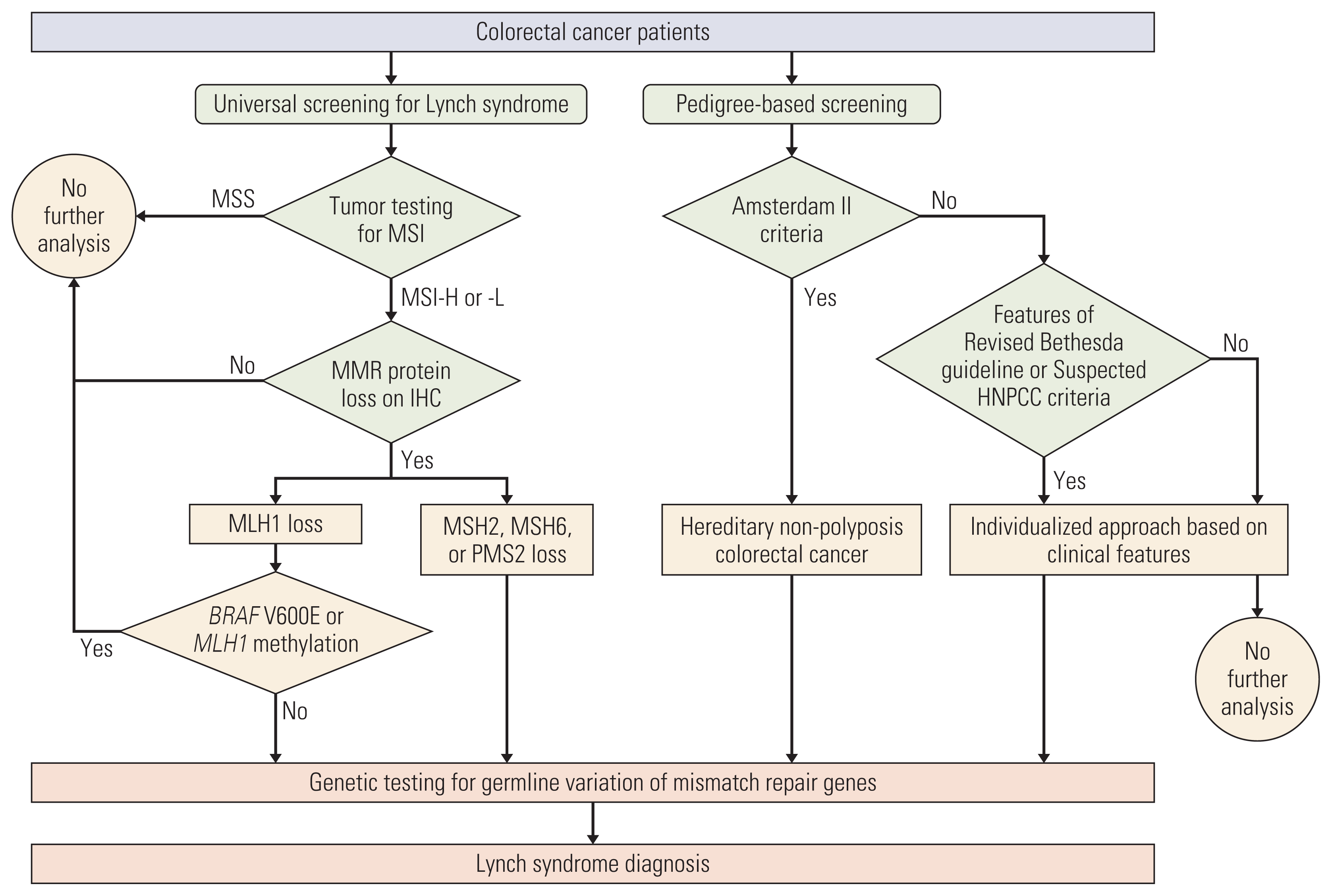
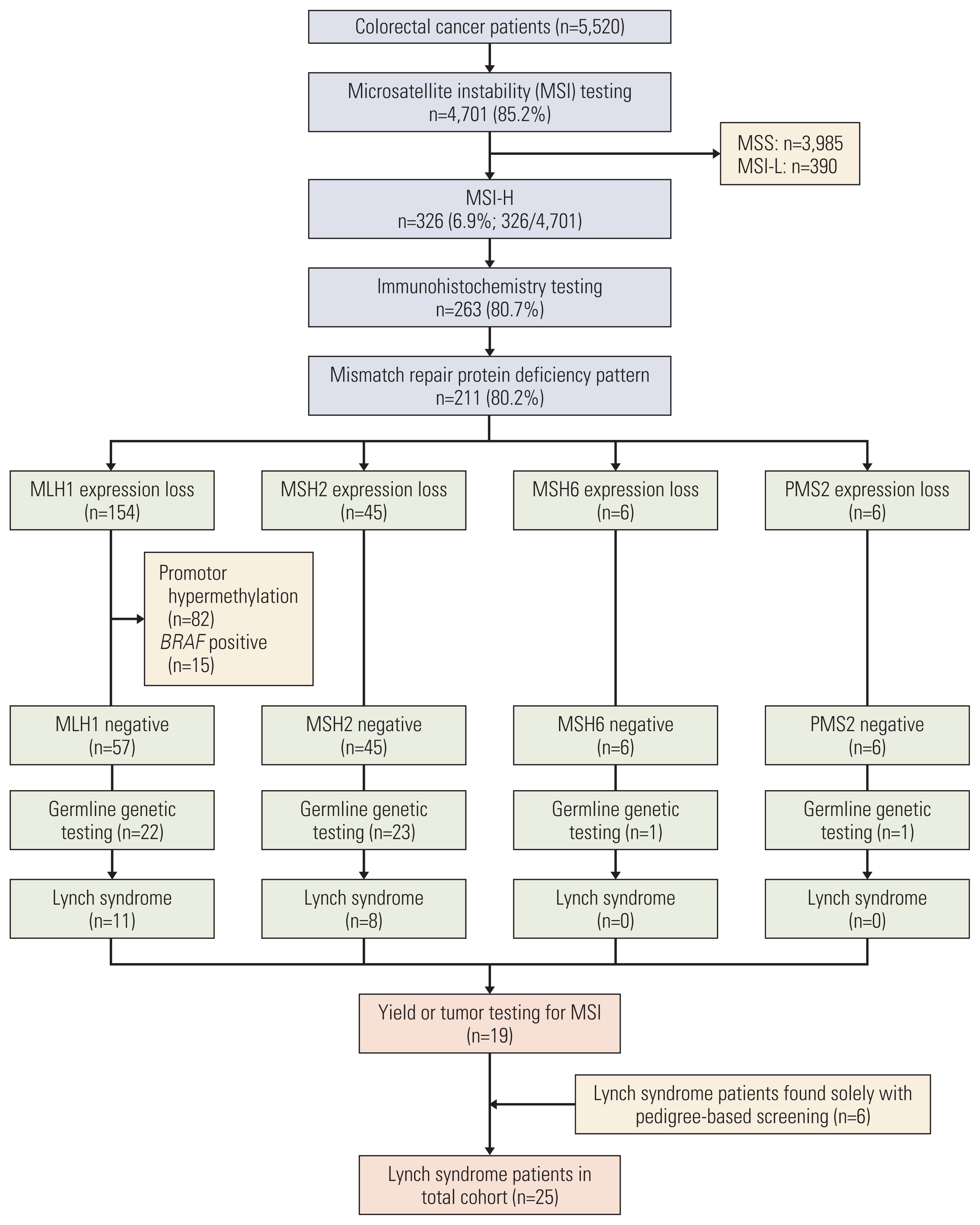
2. Tumor testing for MSI, IHC, and germline genetic testing
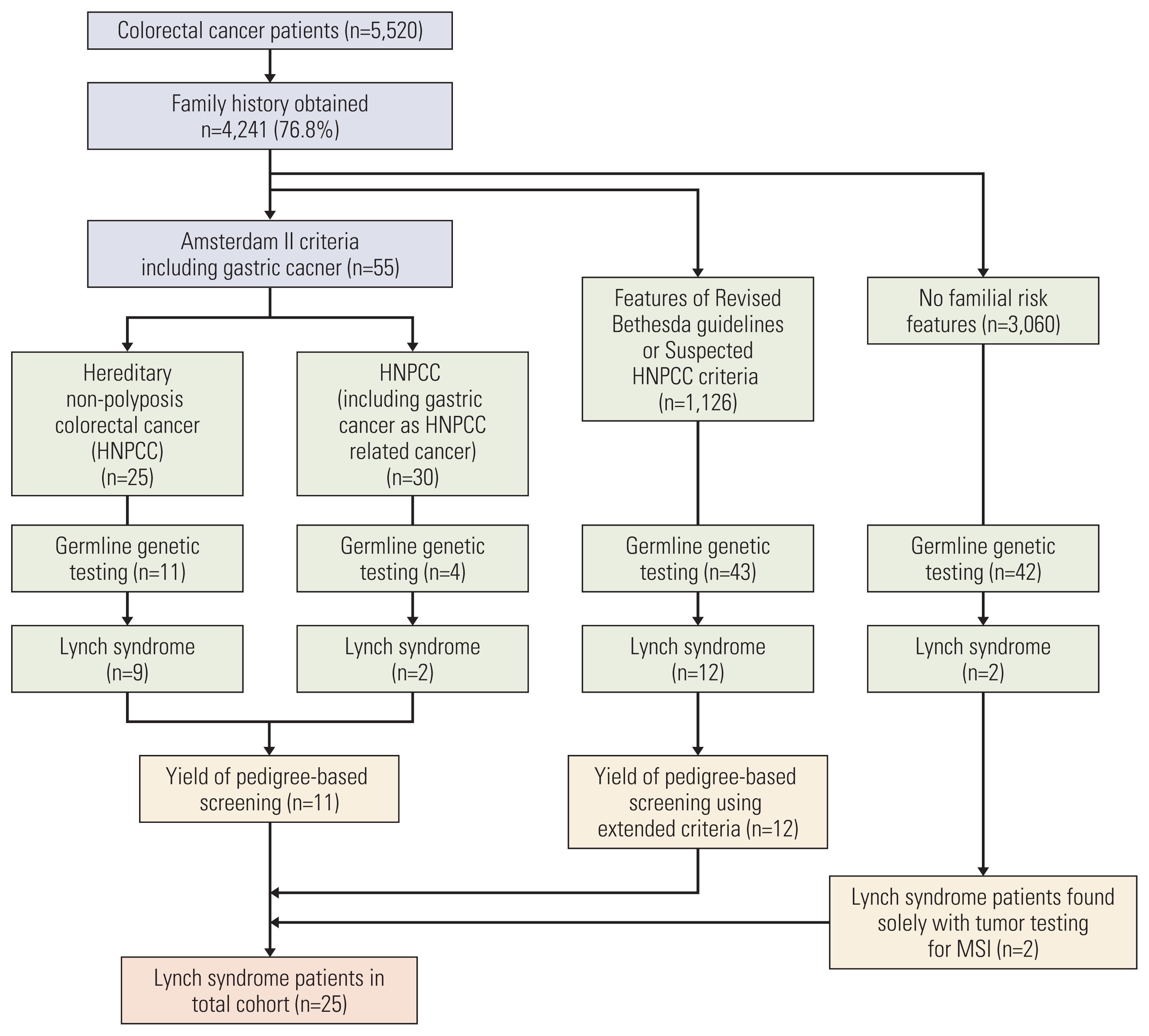
3. Obtaining pedigree and diagnosis of HNPCC
4. Yield analysis
Table 1
| Guideline | Guideline criteria |
|---|---|
| Amsterdam II criteria [16] | There should be at least three relatives with and HNPCC-associated cancer. One should be a first-degree relative of the other two. At least two successive generations should be affected. At least one should be diagnosed before age 50. |
| Revised Bethesda guidelines [18] |
Tumors from individuals should be tested for MSI in the following situations:
|
| Suspected HNPCC criteria [19] | At least two HNPCC-associated cancers in first-degree relatives and |
![]()
Results
1. Patients
Table 2
| No. | Sex/Age | Synchronous/Metachronous cancers, age of diagnosis (yr) | Tumor location | Differentiation | Tumor stage | MSI | IHC | MMR pattern | Mutation gene | Grouped by pedigree analysis | Diagnosed with universal screening | PREMM5 prediction score (%)a) |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | F/29 | Endometrial, 29 | Rectum | WD | 0 | MSI-H | +/nd/nd/nd | Insufficient | MSH2 c.256G>T | HNPCC | No | > 50 |
| 2 | M/40 | Duodenal, 40 | A-colon | WD | 1 | MSI-H | +/−/−/+ | MSH2 | MSH2 c.687dupA | HNPCC | Yes | 44.4 |
| 3 | F/34 | - | A-colon | MD | 2 | MSI-H | +/−/−/+ | MSH2 | MSH2 c.402delT | Suspicious | Yes | 14.6 |
| 4 | M/41 | - | S-colon | MD | 3 | MSI-H | −/+/+/− | MLH1 | MLH1 c.1409+1G>A | Suspicious | Yes | 11.4 |
| 5 | F/37 | - | A-colon | PD | 2 | MSI-H | −/+/+/− | MLH1 | MLH1 c.1039-4_1068del | Suspicious | Yes | 18.3 |
| 6 | F/55 | Gastric, 61 | Multiple | WD | 2 | MSI-H | −/+/+/− | MLH1 | MLH1 c.790+2T>A | Suspicious | Yes | 15.4 |
| 7 | F/44 | - | Multiple | MD | 3 | MSI-H | −/+/+/− | MLH1 | MLH1 c.1409+1G>A | Suspicious | Yes | 5.9 |
| 8 | M/56 | Small bowel sarcoma, 56 | S-colon | WD | 2 | MSI-H | +/+/nd/nd | Insufficient | MLH1 c.1772_1775delATAG | HNPCC | No | 29.9 |
| 9 | F/29 | - | A-colon | MD | 2 | MSI-H | −/+/+/− | MLH1 | MLH1 c.1667+1G>A | HNPCC | Yes | > 50 |
| 10 | F/40 | - | A-colon | MD | 3 | MSI-H | −/+/+/− | MLH1 | MLH1 c.67G>T | Suspicious | Yes | 4.7 |
| 11 | F/49 | T-colon, 52 | A-colon | PD | 2 | MSI-H | +/−/−/+ | MSH2 | MSH2 c.226C>T | Suspicious | Yes | 3.4 |
| 12 | M/50 | - | T-colon | MD | 2 | MSI-H | −/+/+/− | MLH1 | MLH1 c.67G>T | HNPCC | Yes | 30.4 |
| 13 | M/61 | Thyroid, 64 | A-colon | MD | 1 | MSI-H | +/−/−/+ | MSH2 | MSH2 c.1147c>T | Suspicious | Yes | 3.6 |
| 14 | F/61 | Endometrial, 63 | A-colon | WD | 1 | MSS | +/+/+/+ | All positive | MSH2 c.1580delG | Suspicious | No | > 50 |
| 15 | M/52 | - | A-colon | MD | 2 | MSS | +/−/−/+ | MSH2 | MSH2 c.2699C>A | HNPCC | No | > 50 |
| 16 | M/46 | - | A-colon | MD | 2 | MSI-H | +/−/−/+ | MSH2 | MSH2 c.256G>T | HNPCC | Yes | > 50 |
| 17 | M/47 | - | Rectum | WD | 2 | MSI-H | +/−/−/+ | MSH2 | MSH2 c.2089T>C | HNPCC | Yes | 39.7 |
| 18 | F/47 | - | A-colon | MD | 2 | MSI-H | −/+/+/− | MLH1 | MLH1 c.1721T>C | HNPCC | Yes | 22.9 |
| 19 | F/75 | Gastric, 74 & 79; Breast, 77 | A-colon | MD | 1 | MSI-H | −/+/+/− | MLH1 | MLH1 c.1758dupC | No history | Yes | N/A |
| 20 | F/41 | Endometrial, 41 | D-colon | PD | 3 | MSI-H | +/−/−/nd | MSH2 | MSH2 c.1888G>T | Suspicious | Yes | 17.2 |
| 21 | M/23 | - | A-colon | MD | 3 | MSI-H | nd | Not done | MLH1 c.1758dupC | HNPCC | No | > 50 |
| 22 | M/35 | - | S-colon | PD | 3 | MSI-H | +/−/−/+ | MSH2 | MSH2 c.1129C>T | HNPCC | Yes | 47.2 |
| 23 | M/58 | - | Multiple | PD | 3 | MSI-H | −/+/+/− | MLH1 | MLH1 c.1758dupC | No history | Yes | N/A |
| 24 | M/23 | - | D-colon | MD | 4 | MSI-H | nd | Not done | MSH2 c.2653C>T | Suspicious | No | 35.0 |
| 25 | M/20 | - | A-colon | WD | 3 | MSI-H | −/+/+/− | MLH1 | MLH1 c.1721T>C | Suspicious | Yes | > 50 |
A-colon, ascending colon; D-colon, descending colon; HNPCC, hereditary nonpolyposis colorectal cancer; IHC, immunohistochemistry; MD, moderately differentiated; MMR, mismatch repair; MSI, microsatellite instability; MSI-H, microsatellite instability high; MSS, microsatellite stable; N/A, not available; nd, not done; PD, poorly differentiated; PREMM, prediction model for gene mutations; S-colon, sigmoid colon; T-colon, transverse colon; WD, well differentiated.
a) PREMM5 model prediction score was adapted from the Dana Farber Cancer Institute (http://premm.dfci.harvard.edu/).
![]()
 XML Download
XML Download