

 PDF
PDF Citation
Citation Print
Print



Introduction
Materials and Methods
1. Study population
2. Data collection
3. Statistical analyses
Results
1. Patient characteristics
Table 1
| Total | WT | RCC | CCSK | MRTK | CMN | Others | p-value | |
|---|---|---|---|---|---|---|---|---|
| No. (%) | 439 (100) | 342 (77.9) | 36 (8.2) | 24 (5.5) | 16 (3.6) | 12 (2.7) | 9 (2.1) | |
| Male:female | 240:199 | 183:159 | 16:20 | 17:7 | 10:6 | 9:3 | 5:4 | 0.25 |
| Age at Dx (mo) | < 0.001 | |||||||
| Median (range) | 27.1 (0–225.5) | 27.1 (0–225.5) | 128.3 (13.6–214.6) | 20.5 (0–179.9) | 7.3 (1.9–61.6) | 0.2 (0–1.7) | 23.4 (5.5–154.1) | |
| IQR | 13.0–55.3 | 14.1–51.1 | 63.6–182.1 | 12.2–37.0 | 4.7–10.9 | 0.1–0.4 | 10.9–26.4 | |
| Laterality right: left | 216:202 | 164:159 | 21:15 | 13:10 | 8:7 | 4:8 | 6:3 | 0.7945 |
| Bilateral (%) | 21 (4.8) | 19 (5.6) | 0 | 1 (4.2) | 1 (6.2) | 0 | 0 | |
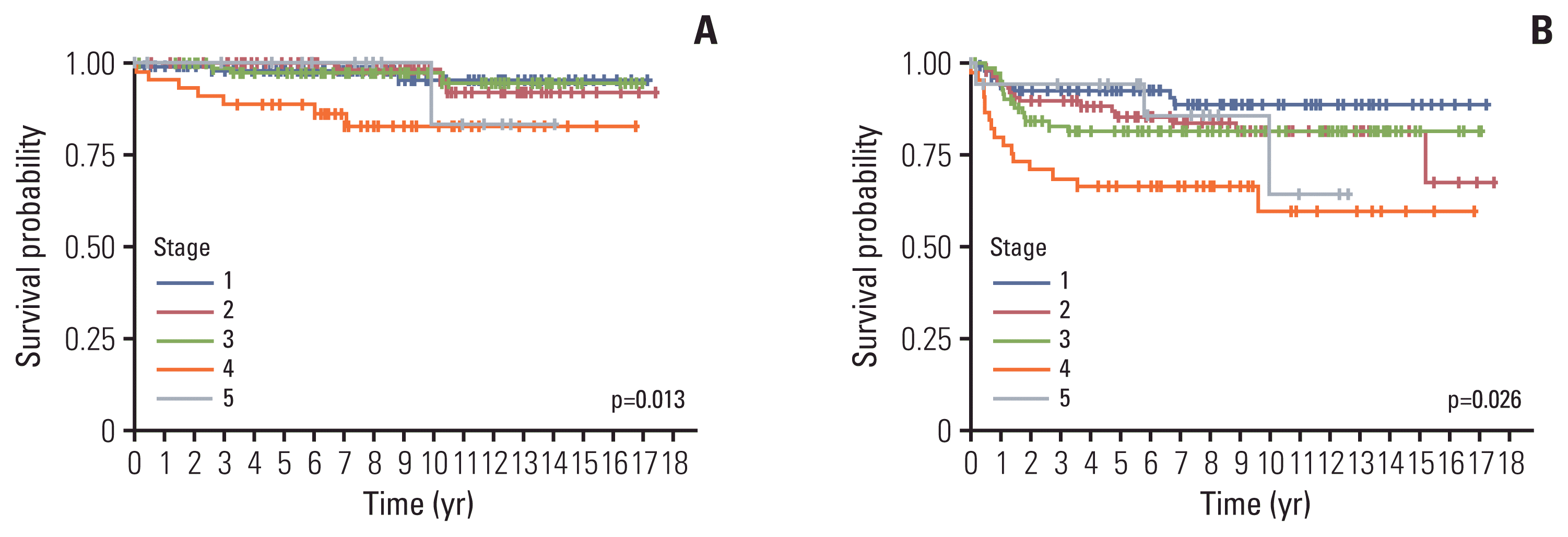
| Stage (%) | ||||||||
| I | 127 (28.9) | 102 (29.8) | 14 (38.9) | 5 (20.8) | 3 (18.8) | 2 (16.7) | 1 (11.1) | |
| II | 108 (24.6) | 89 (26.0) | 5 (13.9) | 7 (29.2) | 5 (31.2) | 0 | 2 (22.2) | |
| III | 104 (23.7) | 84 (24.6) | 9 (25.0) | 6 (25.0) | 2 (12.5) | 0 | 3 (33.3) | |
| IV | 67 (15.3) | 45 (13.2) | 8 (22.2) | 5 (20.8) | 5 (31.3) | 2 (16.7) | 1 (11.1) | |
| V | 20 (4.6) | 19a) (5.6) | 0 | 1 (4.2) | 1a) (6.3) | 0 | 0 | |
| NA | 13 (3.0) | 3 (0.9) | 0 | 0 | 0 | 8 (66.7) | 2 (22.2) | |
![]()
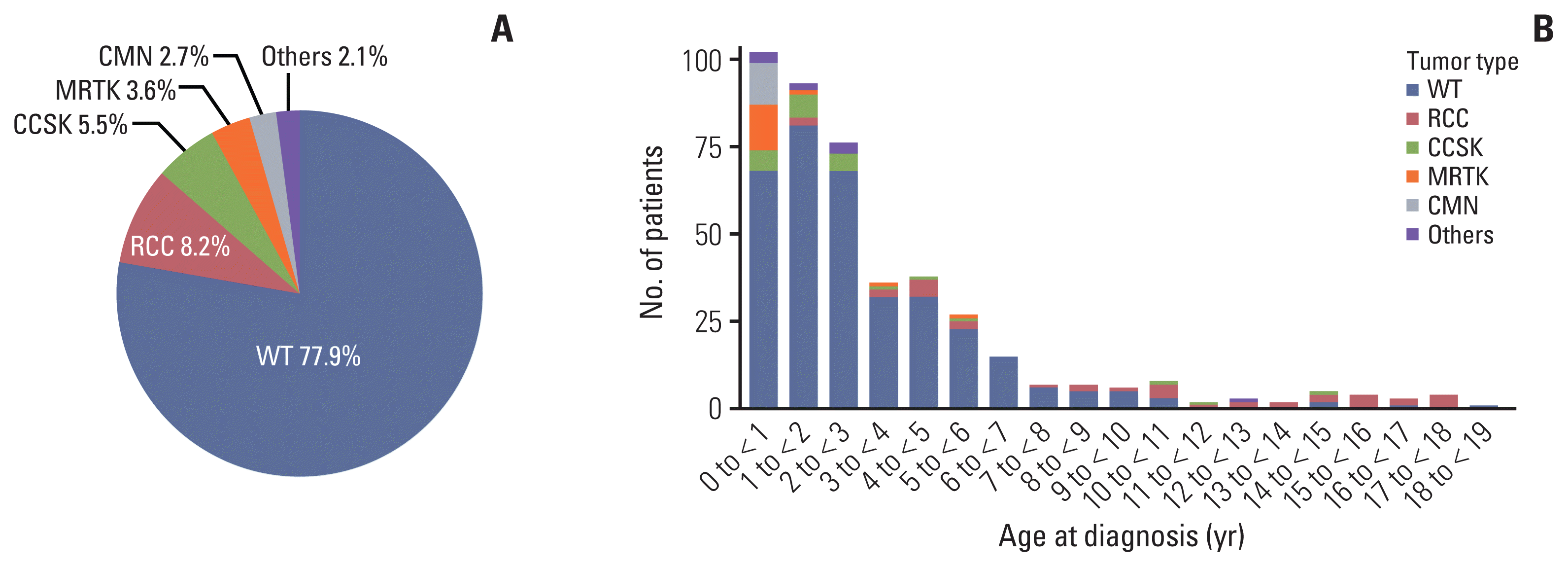
Fig. 2
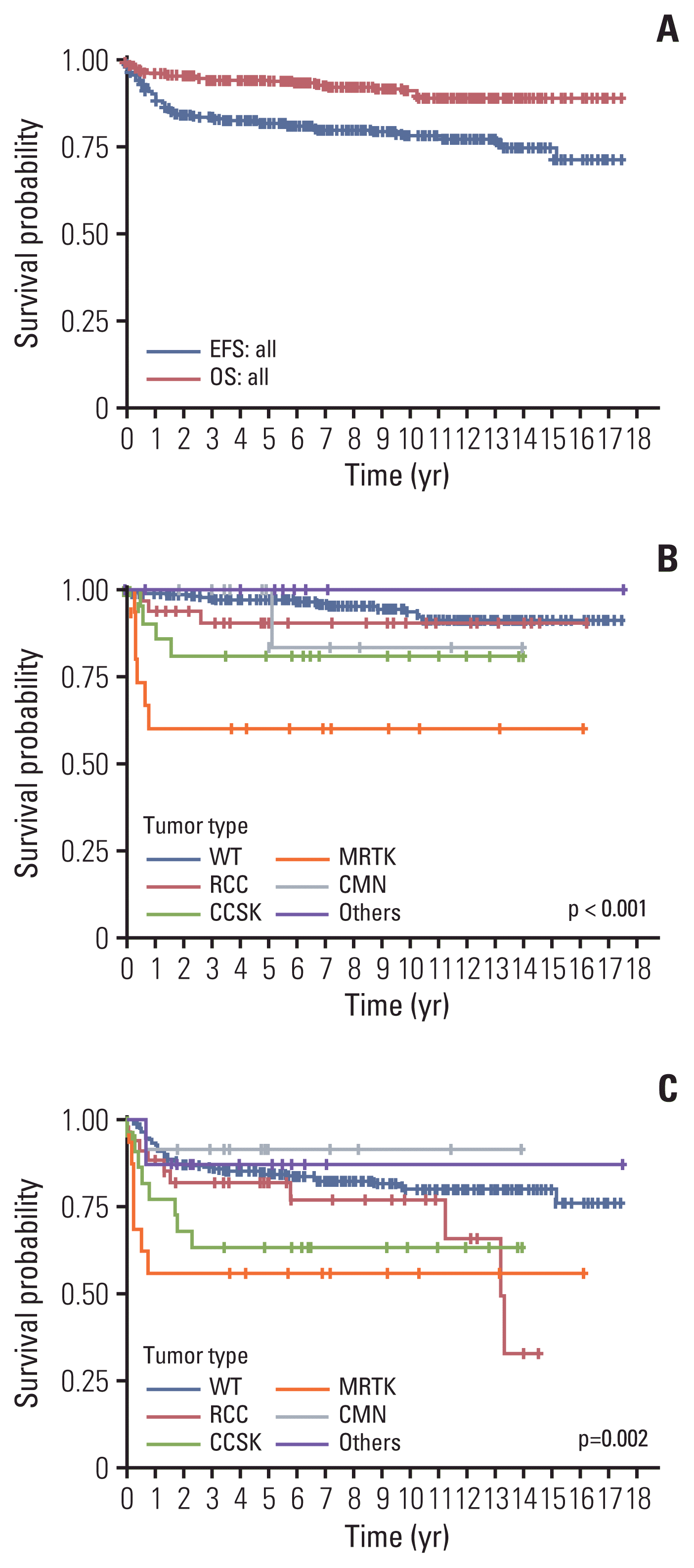
![]()
2. Wilms tumor
1) Stage
Table 2
| No. | Initial Op | Biopsy | Imaging Dx | PreCRx | PostCRx Op | PostCRx | RT | PBSCT (P/R) | Relapse | PD | TRM | SMN | Death | 5-Year OS/EFS (%) | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Overall | 342 | 195 | 126 | 21 | 147 | 144 | 338 | 100 | 6/37 | 45 | 5 | 5 | 3 | 18 | 97.2/84.8 |
| Stage I | 102 | 92 | 6 | 4 | 10 | 9 | 101 | 1 | 0/4 | 8 | 1 | 0 | 0 | 3 | 97.9/93.0 |
| Stage II | 89 | 52 | 32 | 5 | 37 | 37 | 89 | 9 | 0/8 | 11 | 0 | 3 | 0 | 3 | 100/85.6 |
| Stage III | 84 | 28 | 49 | 7 | 56 | 54 | 82 | 50 | 1/11 | 15 | 0 | 0 | 0 | 3 | 97.4/81.6 |
| Stage IV | 45 | 18 | 25 | 2 | 27 | 27 | 44 | 36 | 2/10 | 10 | 3 | 1 | 2 | 7 | 88.9/66.7 |
| Stage V | 19a) | 3 | 13 | 3 | 16 | 16 | 19 | 3 | 3/3 | 0 | 1 | 1 | 1 | 1 | 100/94.4 |
| NA | 3 | 2 | 1 | 0 | 1 | 1 | 3 | 1 | 0/1 | 1 | 0 | 0 | 0 | 1 |
Dx, diagnosis; Initial Op, primary (upfront) nephrectomy at the time of diagnosis; NA, stage not assessable; OS/EFS, overall survival/event-free survival; PBSCT (P/R), high-dose chemotherapy and peripheral blood stem cell transplantation (primary/relapsed); PD, progressive disease; PostCRx, postoperative chemotherapy; PostCRx Op, post-chemotherapy nephrectomy; PreCRx, preoperative chemotherapy; RT, radiation therapy; SMN, second malignant neoplasm; TRM, treatment-related mortality.
![]()
2) Diagnosis and pathology
3) Treatment
4) Outcome
3. Non-Wilms renal tumors
1) Renal cell carcinoma
Table 3
| No. | Initial Op | Biopsy | Imaging Dx | PreCRx | PostCRx Op | PostCRx | RT | PBSCT (P/R)d) | Relapse | PD | TRM | SMN | Death | 5-Year OS/EFS (%) | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| RCC | 36a) | 30 | 6 | 0 | 5 | 3 | 4 | 5 | 2/0 | 6 | 4 | 0 | 1b) | 3 | 90.6/82.1 |
| CCSK | 24c) | 15 | 8 | 1 | 8 | 6 | 20 | 16 | 1/1 | 5 | 3 | 0 | 0 | 4 | 81.1/63.6 |
| MRTK | 16 | 10 | 5 | 1 | 6 | 13 | 12 | 8 | 5/0 | 2 | 4 | 0 | 0 | 6 | 60.3/56.2 |
| CMN | 12 | 12 | 0 | 0 | 0 | 12 | 4 | 1 | 0 | 1 | 0 | 0 | 0 | 1 | 100/91.7 |
| Others | 9 | 4 | 4 | 1 | 5 | 9 | 8 | 1 | 1/0 | 1 | 0 | 0 | 0 | 0 | 100/87.5 |
CCSK, clear cell sarcoma of the kidney; CMN, congenital mesoblastic nephroma; Dx, diagnosis; Initial Op, primary (upfront) nephrectomy at the time of diagnosis; MRTK, malignant rhabdoid tumor of the kidney; OS/EFS, overall survival/event-free survival; PBSCT (P/R), allogeneic or autologous peripheral blood stem cell transplantation (primary/relapsed); PreCRx, preoperative chemotherapy; PD, progressive disease; PostCRx, postoperative chemotherapy; PostCRx Op, post-chemotherapy nephrectomy or nephrectomy immediately after biopsy; PreCRx, preoperative chemotherapy; RCC, renal cell carcinoma; RT, radiation therapy; SMN, second malignant neoplasm; TRM, treatment-related mortality; WT, Wilms tumor.
![]()
 XML Download
XML Download