

 PDF
PDF Citation
Citation Print
Print



서론
네말린 근병증은 선천성 근병증 중 가장 흔한 질환 중 하나로, 병리학적으로 골격근 섬유에 네말린 로드(nemaline rod)라는 특징적인 소견이 관찰 된다[1]. 이는 유전적 이질성을 가지며 선천성 근병증의 17%를 차지하고, 대략 1/50,000의 확률로 태어난다[2]. 전형적으로 나타나는 임상증상은 근육 약화, 근육 긴장성 손상, 심부건 반사의 소실 등이다. 근육 약화는 일반적으로 얼굴, 목 굴곡근, 사지 근위부 근육에서 주로 발생한다. 얼굴 근육이 심하게 약해지며, 마르고 턱이 두드러져 보이는 특징적인 얼굴 모양이 나타나게 된다. 또한 호흡근이나 저작근이 약해져서 호흡 장애와 연하 장애가 발생하며, 호흡 장애가 사망의 가장 큰 원인이 된다[3]. 1999년도 European Neuromuscular Centre Internetional Consortium (ENMC)에서는 네말린 근병증을 발생 연령 및 임상증상의 중증도에 따라서 6가지 타입으로 다음과 같이 분류하였고, 이는 (1) 극심한 선천성 형, (2) 아미쉬 형, (3) 중간 선천성 형, (4) 전형적 선천성 형, (5) 소아기 발병 형, (6) 성인기 발병 형이다. 네말린 근병증은 골격근의 얇은 필라멘트를 구성하는 단백질들을 생성하는 유전자에 돌연변이가 발생하는 것으로, 단백질에는 네뷸린, 트로포마이신 베타와 감마, 액틴, 트로포닌 티가 있으며, 이들의 연결은 Kelch-like family member 40으로 안정화 되어 있다. 네말린 근병증의 원인 돌연변이가 되는 유전자는 대표적으로 NEB, ACTA1, TPM3, TPM2, TNNT1, CFL2, KBTBD13, KLHL40, KLHL41, KBTBD13가 있다[1,4].
NEB는 네뷸린 단백질을 형성하는 유전자로, NEB에 발생하는 돌연변이는 상염색체 열성 유전이며 네말린 근병증에서 약 50% 정도의 증례를 보이는 돌연변이고[2], 우리나라에서도 가장 흔한 병적 변이로 알려져 있다[1]. NEB 돌연변이에서 발생하는 가장 흔한 임상증상에는 원위부 근육 약화가 있다[5].
KLHL40은 Kelch-like family member 40 단백질을 형성하는 유전자로, KLHL40 돌연변이 또한 상염색체 열성 유전이며 임상적으로 극심한 네말린 근병증을 일으키는 대표적인 돌연변이이며, Ravenscroft 등[6]은 약 19.6%로 발생한다고 보고하였다. 특히 이 표현형을 가지는 유형의 28%는 일본에서 대부분 발견되고 있으며[4], 대부분 태동의 감소, 관절 구축, 호흡 부전, 연하 장애를 태어날 때부터 가지게 된다. 선천적인 골절도 흔하게 나타나며 환아의 절반 이상이 중력에 반하는 힘을 주지 못한다. 평균 수명은 약 5개월이라고 알려져 있다. Ravenscroft 등[6]은 c.1582G>A 변이가 일본에서 주로 발생하는 돌연변이이며 이는 한국, 터키에서도 정의된다 밝혔다.
본 증례에서는 출생 직후 심한 전신 근육 약화를 보인 신생아에서 차세대 염기서열분석법(next generation sequencing, NGS)을 이용한 유전자 패널검사를 통해 네말린 근병증을 진단한 경험을 보고하고자 한다.
증례
재태 연령 37주 5일에 출생 체중 3,500 g, 제왕절개로 아프가 점수 1분 5점, 5분 7점으로 출생한 여아가 출생 후 잘 울지 않고 청색증이 동반되어 출생 당일 본원으로 전원 되었다. 환아의 어머니는 임신으로 인한 고혈압 이외 특이 병력 없었고, 산전 진찰상에서도 이상 소견 없었으며 출생 전 태동 감소 소견을 보이지 않았다. 환아는 첫째이며, 유전 질환의 가족력은 없었고 내원 당시 키 46 cm (10 백분위수), 체중 3,290 g (50 백분위수), 두위 38 cm (90 백분위수), 태변 착색이 있었으며 전반적 근긴장도 저하가 있었다. 이마가 돌출되어 있으며, 이마에 포도주색 반점이 있고, 평평한 얼굴, 양안 격리증, 하악 발육 부전, 인중 모양이 짧고, 흉부 함몰을 동반한 술통 가슴, 목이 짧고 익상경이 있는 모습을 보였다. 또 모든 관절이 느슨하게 늘어져 있고, 각 관절의 구축이 있었으며, 손가락이 비정상적으로 길고 얇은 형태이며 등과 다리에는 다모증이 있었다. 선천적 골절은 존재하지 않았다.
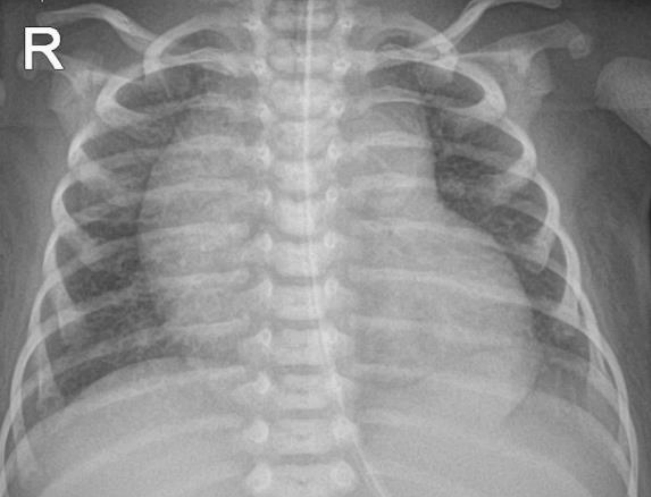
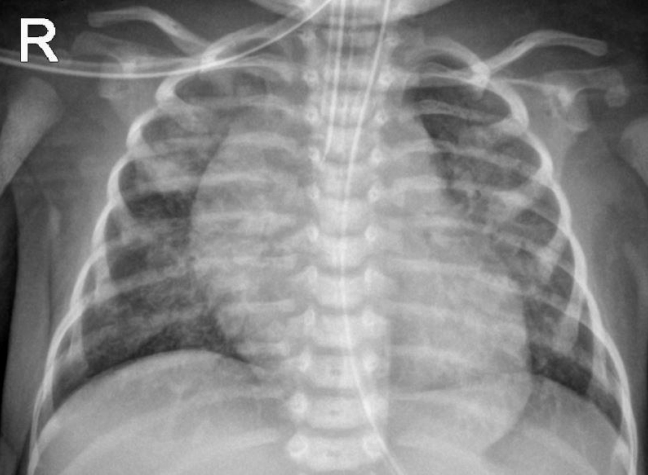
환아는 처음 내원 당시 혈액가스검사에서 pH 7.357, pCO2 41.1 mm Hg, BE –2.5 mmol/L, HCO3 22.4 mmol/L 소견으로 호흡성 산증은 없었고, 젖산 2.57 mmol/L (0.7–2), CK 294 U/L (26–192), CKMB 18.3 μg/L (0.0–5.0), pro-BNP 36,377 pg/mL (0–320)로 근육 효소 및 심장 효소 수치가 상승하였다. 내원 당시에는, 고유량 비강 캐뉼라 산소 주입으로 산소포화도 90%–100%로 유지되었으나, 단순 흉부방사선사진에서 심비대 및 폐렴 악화 소견 보이고 자발 호흡이 힘들어지면서 기관 삽관하여 기계 환기 치료를 시행하였고 또한 연하 장애도 동반되어 있어, 위관 삽입하여 식이를 진행하였다(Figures 1, 2). 호흡 곤란 및 연하 장애, 근긴장도 저하, 특징적 외형으로 다발성 기형 감별을 위하여 뇌 초음파, 복부 초음파를 시행하였으며 정상이었다. 다양한 선천적 기형에 대한 유전학적 원인을 규명하기 위해 고식적 핵형 분석 및 염색체 마이크로어레이 검사를 시행하였으며, 선천성 신경근 질환을 감별하기 위해 선천성 대사이상 검사, SMN1 (척수근위축증), GAA (폼페병) 유전자검사를 시행하였다.
핵형은 46, XX로 정상이었고 염색체 마이크로어레이 검사에서도 임상적으로 중요한 염색체의 미세 결실, 중복은 발견되지 않았다. 선천성 대사이상 선별검사는 정상이었고, 긴 사슬 지방산 검사에서도 C22:0 28.414 μmol/L (0.0–96.3), C24:0 21.136 μmol/L (0.0–91.4), C26:0 0.266 μmol/L (0.0–1.31)로 정상이었다. SMN1, GAA 유전자 검사에서도 병적 변이는 발견되지 않았다.
이외에 다양한 유전성 근질환을 감별하기 위해 NGS를 이용한 유전자 패널검사를 시행하였다. 이를 통해 KLHL40 유전자에서 2개의 병적 변이(pathogenic variant)가 발견되었다: KLHL40 c.[1405G>T]; [1582G>A] (p.[Gly469cys];[Glu528Lys]) (Figure 3). 또한 부모검사에서 복합 이형접합성이 확인되어, 환아는 네말린 근병증으로 진단 되었다.
환아는 입원 치료 중 심비대를 유발한 3.2 mm 크기의 동맥관개존은 자연적으로 폐쇄되었으며, 폐렴 치료로는 항생제 암피실린-설박탐 제제를 2주간 사용하였다. 폐렴 호전 이후 기계 환기 이탈을 시도하였으나 지속적으로 무호흡이 발생하여, 카페인(neocaf)을 사용하면서 기계 환기 치료를 유지하였고, 이후 생후 19일째 기계 환기를 이탈하였다. 하지만 객담 배출이 원활하게 이루어지지 않았고, 식이 증량 시 분비물이 증가하면서 산소포화도가 떨어지는 경우가 발생하여, 지속적 객담 흡입 및 비강 캐뉼라 산소를 유지하였고, 위관 식이를 지속하였다. 또 눈동자의 움직임이 없고, 양안을 완전히 뜨고 감지 못하는 상태였지만 대광 반사는 정상적으로 이루어졌으며, 이외 소견은 정상이었다. 생후 6일째부터 혈액가스검사상에서 pH 7.674, pCO2 40.5 mm Hg, BE 26.7 mmol/L, HCO3 49.3 mmol/L로 구토 및 이뇨제 사용 없이 대사성 알칼리증이 발생하였고, 저칼륨혈증이 동반되어 시행한 혈액, 소변, 아미노산 및 유기산 검사는 정상이었다. 대사성 알칼리증에 대하여 경구 칼륨 복용하면서 경과 관찰하였다. 이후 보호자 요구로 재활 및 추가적 치료를 위해 상급병원으로 전원 되었다.
고찰
네말린 근병증은 발생하는 연령에 따라 다양한 임상증상을 나타내는데, 산전과 신생아기에 발병되는 네말린 근병증의 경우 호흡 부전을 보이기에 가장 예후가 좋지 않다[1,7]. 본 증례는 임상증상과 발병 시기를 고려하였을 시, ENMC 기준에 따라 극심한 선천성 형에 해당된다. 혈액검사에서 CK는 정상이거나 약간 상승되어 있고, 이는 근육의 손상을 의미하며 환아의 경우도 294 U/L (26–192)로 상승 소견을 관찰할 수 있었으나 이는 네말린 병증을 진단할 수 있는 요소는 아니다. 현재까지는 진단을 위해서는 관절 구축, 호흡 장애, 연하 장애 등의 임상증상을 통해서 진단을 유추하고, 근전도검사, 조직검사를 시행한 이후 유전학적 검사를 진행 하였다[8]. 하지만 환아는 기계 환기 치료 중인 상태였기에 근전도검사 및 근육 조직검사를 시행하지 못하였다. 따라서 확진을 위해 염기서열 분석 등의 유전학적 검사법의 도움을 받았고, 이 증례에서는 유전적 근육퇴행위축 질환의 유전자 변이를 알아보기 위하여 NGS를 이용하였다. 이전 연구에 따르면 Lee 등[1]과 Park 등[9]은 임상증상과 근조직검사에서 네말린 근병증으로 진단받은 환자들을 대상으로 하여 전체 엑솜 염기서열 분석법(whole exome sequencing, WES) 및 Sanger 염기서열 분석법(Sanger sequencing)을 사용하였다. 그러나 Moreau-Le Lan 등[2]과 Piga 등[10]은 네말린 근병증은 유전적 이질성을 가진 질환이고, NEB 유전자가 183개의 엑손을 가지고 있는 크고 복잡한 유전자라는 구조 특성상 기존의 염기서열 분석법을 사용할 경우 시간 소비적이며 비용적으로 비싸고, 근병증 관련 새로운 유전자들이 증가하고 있는 추세로 엑솜 염기서열 분석법 및 Sanger 염기서열 분석법 보다 NGS가 더 효율적이라고 밝히고 있다. Lee 등[1]에서는 NEB 유전자의 경우 엑손 82에서 105까지 반복되는 구간이 있어 정확한 길이를 읽는 데에 WES는 제한이 되지만 NGS를 이용하였을 시, 이 제한점을 극복 할 수 있다고 기술하였다. 본 증례에서는 신생아기에 발병되는 네말린 근병증의 대표적인 증상을 보였고, NGS를 통하여 다음과 같은 유전자검사를 시행하였고 이를 통해 KLHL40 병적 변이를 발견하였다: ACTA1, ATP2A1, BAG3, BIN1, CCDC78, CFL2, CLCN1, CNTN1, CRYAB, DAG1, DOK7, DYNC1H1, FHL1, FLNC, GAA, GNE, IGHMBP2, ITGA7, KBTBD13, KLHL40, KLHL41, LOB3, MTM1, MYH7, NEB, PABPN1, POLG, RYR1, SGCE, SYNE1, TNNT1, TPM2, TPM3, TRIM32, TRPV4, VCP, VPS13A, VRK1. 본 증례에서 보인 KLHL40 유전자의 c.1405G>T (p. Gly469Cys) 변이는 일반 인구집단에서 보고된 바 없는 매우 드문 변이이며, 가상 예측에서 단백질 기능이 손실되었다고 예측되며 ClinVar에서 병적 변이로 분류되어 있다(PMID: 23746549, 25721947). c.1582G>A (p. Glu528Lys) 변이는 전체 인구집단에서 경미한 대립 유전자 빈도(minor allele frequency)로써 0.0061% 확률로 매우 드물게 보고되며, 한국인 인구집단에서는 0.36%인 변이로 가상 예측에서 단백질 기능이 손실되었다고 예측되었고, ClinVar에서 병적 변이로 등록되어 있다(PMID: 23746549). Lee 등[1]과 Park 등[9]의 연구에서 출생 시 증상이 있어도 최종 진단되는 데 걸린 시간은 3개월이 걸렸으나, 본증례의 경우 NGS를 확진 검사로 시행함으로써 최종 진단까지 1개월의 시간이 걸렸다. 이처럼 빠른 확진을 통하여 질병의 예후를 예측하고, 치료 계획 수립에 큰 도움이 되었으며 추후 부모의 유전 상담을 진행할 수 있었다. 또한 이전에는 근조직검사와 같은 침습적인 방법으로 확진할 수 밖에 없었으나 본 증례처럼 최근에는 침습적 방법 없이도, 유전적 검사를 통하여 선천성 근병증을 쉽게 확진 할 수 있다는 점에서도 이 증례는 의의가 있다.
본 증례에서 확인된 KLHL40의 c.1405G>T (p. Gly469Cys) 변이에 대해 일본에서 증례 보고가 있으며, 미스센스 돌연변이(missense mutation)로 인해 단백질이 불안정화되어 네말린 근병증이 발생하게 되며[6], Kawase 등[8]에 의하면 본 증례처럼 태어나자마자 청색증, 호흡 곤란, 전반적 근긴장도 저하가 있었으며, 본 증례와는 다르게 태아 수종(폐 흉수, 전신부종)이 있었다. 근조직검사 시행 후 유전학적 검사를 시행하였으며 c.1405G>T (p. Gly469Cys)의 동형 접합변이를 확인하였다. 일본에서 네말린 근병증을 일으키는 가장 흔한 KLHL40의 변이는 c.1582G>A (p. Glu528Lys)로 알려져 있으나, c.1582G>A (p. Glu528Lys) 복합 이형 접합성이 있는 경우가 없는 경우에 비해 임상증상이 양호하다고 밝혔다6). 본 증례는 복합 이형접합으로 c.1582G>A (p. Glu528Lys) 변이를 가지고 있어 Kawase 등[8]의 증례와 비교할 시 기관 삽관을 9개월까지 지속하여 기관 절개를 시행한 경우에 비해, 빠르게 기계 환기 이탈을 하였다. Kawase 등[8]에서는 외관상의 특이 소견은 보이지 않았으나, 본 증례에서는 양안 격리증, 짧은 인중, 하악 발육 부전, 긴 손가락/발가락 등의 외관상 특징이 있었다. 또한, 우측 위팔뼈와 양측 대퇴골의 골절이 Kawase 등[8]에서는 발견되었으나, 본 증례에서는 골절 소견은 보이지 않았다. 인도의 증례 발표에 의하면 KLHL40 돌연변이에 의한 네말린 근병증에서 구개 파열이 발견되었다고 한다[11].
우리나라의 경우, 네말린 근병증 환자의 33.3%가 NEB 돌연변이였으며, 44.7%가 소아기 발병 형 임이 확인 되었다[1,7]. 신생아기 발병에서는 호흡곤란, 근긴장 저하로 기계 환기를 유지하였으나, 소아기 발병 형의 경우 걸음 장애가 가장 흔한 증상으로 나타났다. Lee 등[1]과 Park 등[9]의 국내 연구에서는 NEB 이외 TPM3, ACTA1의 돌연변이는 발견되었으나, KLHL40 돌연변이는 발견되지 않았다.
최근 KLHL40 돌연변이 네말린 근병증 치료에 있어 아세틸콜린 에스테라아제 억제제가 효과적인 반응을 보였다는 연구 결과에 따라 현재 가는 필라멘트 및 그들 사이의 연결 단백질을 타깃으로 하여 약물 연구가 동물 모델에서 진행 중이나 아직 구체적인 연구결과는 없다[12].
결국 네말린 근병증은 다양한 유전자 돌연변이가 상염색체 우성 및 열성으로 유전되는 질환으로 정확한 분자 유전학적 진단을 통해 환아 및 가족에게 질환에 대한 정보 및 예방, 조기 치료를 제공하는 유전 상담이 중요하다. 그러므로 본 증례처럼 정확하고 빠른 진단을 위해 우선적으로 NGS를 이용하는 것을 고려해 볼 수 있겠다.

 XML Download
XML Download