

 PDF
PDF Citation
Citation Print
Print



INTRODUCTION
Wolff-Parkinson-White (WPW) syndrome is rare among neonates; however, the first manifestation may be shock, which requires immediate attention. When supraventricular tachycardia (SVT) occurs in neonates, symptoms of acute heart failure may appear, manifesting as reduced oral intake, lethargy, and breathing difficulty. If cardiovascular collapse is sustained for a long time, severe metabolic acidosis may occur. We report the case of a healthy 28-day-old female full-term neonate diagnosed with WPW syndrome after cardiogenic shock with non-specific symptoms and SVT. This case report alerts pediatricians to exercise a high index of suspicion for arrhythmia, which is a critical cause of cardiogenic shock, and emphasizes the importance of evaluating suspected cases with serial electrocardiography (ECG).
Go to : 
CASE REPORT
A previously healthy 28-day-old female was brought to the emergency room due to poor feeding. She was born at 39 weeks’ gestation via spontaneous vaginal delivery with a birth weight of 3,020 g. She had visited the local clinic 3 days earlier. Echocardiography was performed at the local clinic to evaluate a gallop rhythm and cardiac murmur, but no abnormalities were found. However, her poor feeding and lethargy worsened the day before she visited our hospital. She did not present with any other signs. Her vital signs were as follows: heart rate (HR) of 160 beats per minute (bpm), respiratory rate (RR) of 30 breaths/min, oxygen saturation of 98% on room air, and body temperature (BT) of 36.6 °C. Initial laboratory tests showed hypoglycemia (serum glucose, 42 mg/dL) and metabolic acidosis (venous blood gas analysis [VBGA]: pH, 7.41; PCO2, 24 kPa; bicarbonate, 15.5 mmol/L; and base excess, –6.5 mmol/L). Initial chest radiograph showed mild cardiomegaly without pulmonary edema (Figure 1). To correct this, a bolus of dextrose was administered and fluid resuscitation was started. Despite these measures, she became lethargic and showed cyanosis 5 hours later and developed hypothermia and tachycardia (HR, 220 bpm; blood pressure, 93/71 mm Hg; RR, 50 breaths/min; oxygen saturation, 78%; and BT 33°C) and severe metabolic acidosis (VBGA, pH, 6.84; PCO2, 25 kPa; bicarbonate, 1.6 mmol/L; and base excess, –28.9 mmol/L). Other laboratory findings included white blood cell count of 15.1×103/μL (35.0% neutrophils, 54.0% lymphocytes, and 11.0% monocytes), and hemoglobin, C-reactive protein, aspartate aminotransferase, alanine aminotransferase, total bilirubin, direct bilirubin, creatine kinase-MB, troponin-T, troponin-I, N-terminal pro-B type natriuretic peptide, and ammonia levels of 14.4 g/dL, 0.79 mg/dL (reference, <0.5 mg/dL), 259 U/L, 193 U/L, 3.5 mg/dL, 1.1 mg/dL, 27.7 ng/mL (reference range, 0.1 to 5.8 ng/mL), 0.208 ng/mL (reference range, <0.1 ng/mL), 0.52 ng/mL (reference range, <0.16 ng/mL), 45,351.0 pg/mL (reference range, <84 pg/mL), and 323 μg/dL, respectively. Moreover, disseminated intravascular coagulation (DIC) was identified (platelet count 58×103/μL; prothrombin time international normalized ratio, 4.96; activated partial thromboplastin time, 56.2 seconds; fibrinogen <50 mg/dL; figrin (ogen) degradation product, 116.2 μg/mL; D-dimer, 76.6 mg/L). She was admitted to the neonatal intensive care unit for further evaluation. Despite additional fluid resuscitation with high-dose dextrose infusion and oxygen administration via a high-flow nasal cannula, hypoxemia persisted and tachycardia worsened shortly after admission. We immediately performed endotracheal intubation and applied mechanical ventilation. Tachycardia was observed (HR of 240 bpm) on an ECG monitor. A diagnosis of SVT was made, and adenosine was administered immediately. Afterward, her ECG showed a normal sinus rhythm with an HR of 170 bpm, and her dyspnea and lethargy improved. After she became stable, 12-lead ECG was performed, which revealed normal sinus rhythm without ventricular pre-excitation (Figure 2A). Echocardiography showed mild left ventricular dysfunction with mild mitral and tricuspid regurgitation. After she was stabilized, metabolic acidosis gradually improved while maintaining high-dose dextrose and high caloric feeding until completely excluded.
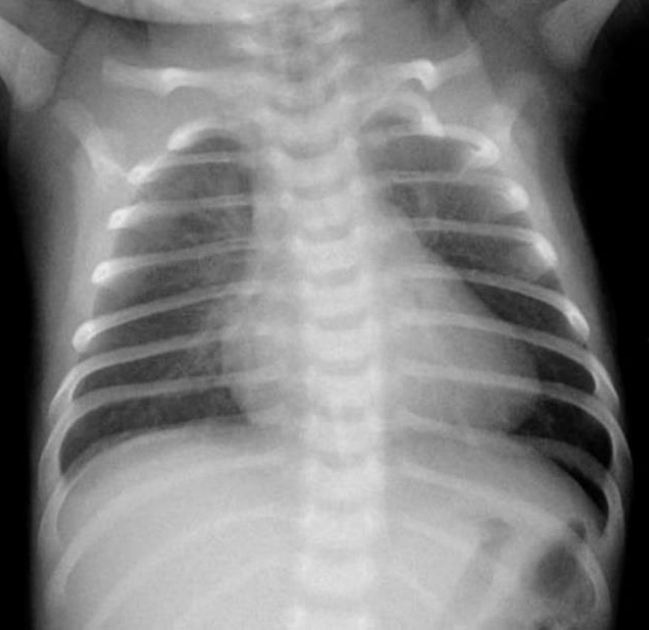 | Figure 1.Chest radiograph of a 28-day-old female with cardiovascular collapse shows mild cardiomegaly (cardiothoracic ratio 56%) without pulmonary edema.
|
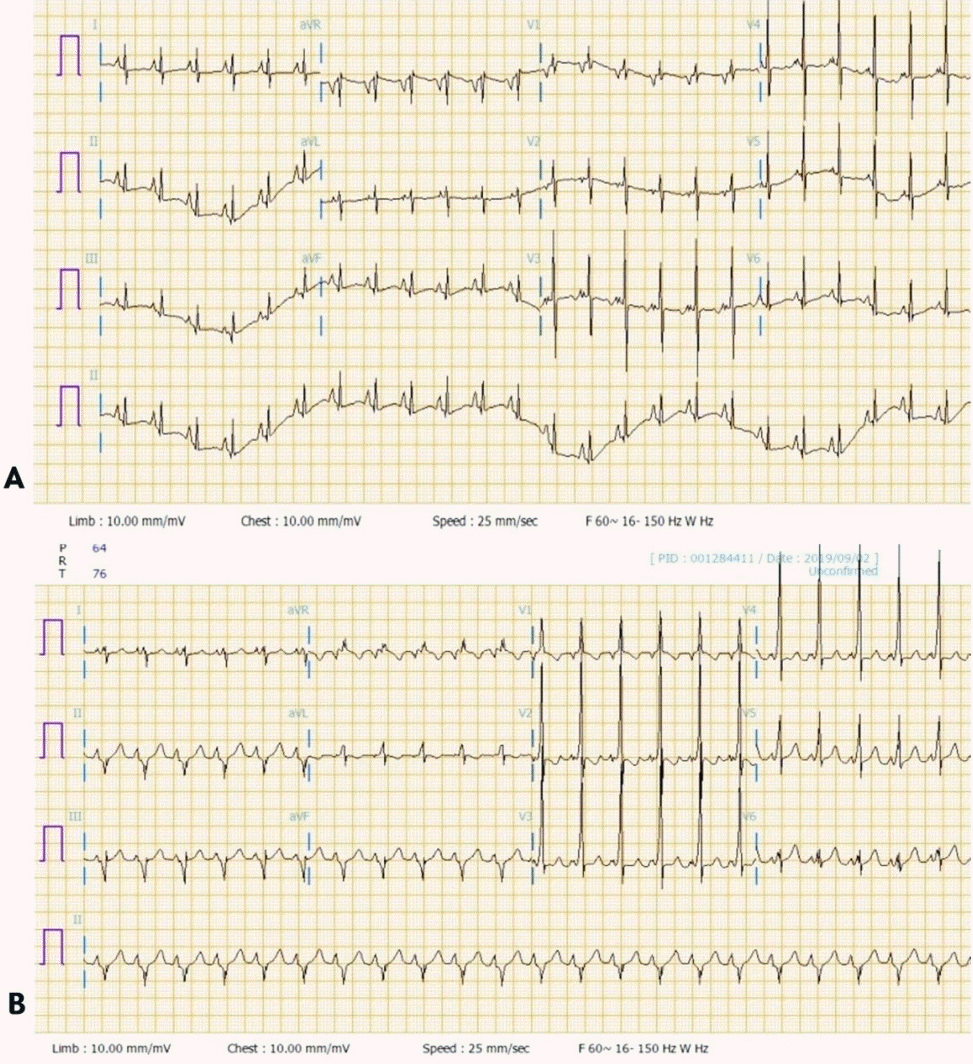 | Figure 2.(A) Electrocardiography tracing of a 28-day-old female with cardiovascular collapse. Electrocardiography after the first episode of supraventricular tachycardia shows normal sinus rhythm (heart rate of 150 beats per minute). (B) Electrocardiography after the second episode of supraventricular tachycardia shows a short PR interval and wide QRS complex in all leads. This indicates ventricular pre-excitation (delta wave) resulting in the diagnosis of Wolff-Parkinson-White syndrome.
|
We needed to find possible causes of sudden collapse and severe metabolic acidosis. An inborn error of metabolism was suspected because she had hypoketotic hypoglycemia, hyperammonemia, mildly elevated liver transaminases, and severe metabolic acidosis. However, newborn screening by tandem mass spectrometry and levels of plasma amino acid, urine organic acid, plasma acylcarnitine, and total and free carnitine suggested that a fatty acid oxidation disorder was unlikely. Furthermore, sepsis was considered because of the systemic inflammatory response showing hypothermia, tachypnea, tachycardia, metabolic acidosis, and DIC. However, there was no obvious increase in the level of inflammatory markers. Additionally, no organism was detected in blood culture, urine culture, and multiplex polymerase chain reaction performed on a nasopharyngeal specimen. Thus, cerebrospinal fluid testing was not performed.
On hospitalization day 8, she suddenly developed tachycardia and desaturation. The ECG monitor revealed SVT. Adenosine was administered immediately, and ECG tracing returned to normal sinus rhythm. A subsequent 12-lead ECG showed ventricular pre-excitation diagnostic of WPW syndrome (Figure 2B). Propranolol was started as maintenance treatment of SVT with WPW syndrome. Subsequently, she was discharged on hospitalization day 25 without any further events.
A novel sequence variant, c.238A>G (p.Thr80Ala), in the myosin light chain 2 (MYL2) gene that is related to hypertrophic cardiomyopathy (HCM) and conduction defects on a nextgeneration sequencing-based panel test was found after discharge. Additional parental genetic tests confirmed that the father had the same mutation, but he did not show cardiac symptoms. Further evaluation will be performed on him. The patient is now 16 months old and still receiving beta-blocker treatment in the outpatient clinic. Ventricular pre-excitation remains evident in the follow-up ECG; however, echocardiography revealed normalized findings.
Go to :
DISCUSSION
Hypovolemia, bacterial sepsis, and congenital heart disease are common causes of acute cardiovascular collapse in young infants. SVT is the most common tachyarrhythmia in the pediatric population. In addition, WPW syndrome is rare, with an incidence rate of 0.1% to 0.2% among neonates; however, the first manifestation may be shock, requiring immediate attention [1,2]. Furthermore, 50% of young patients with WPW syndrome with cardiac arrest had no previous cardiac symptoms [1,2]. Depending on the duration of SVT, symptoms can range from asymptomatic presentation to cardiogenic shock. Poor feeding, lethargy, and irritability may be present with prolonged tachyarrhythmia. These symptoms appear non-specific. Therefore, it can be mistaken for anemia, fever, sepsis, dehydration, and congenital structural cardiac defects [3]. In severe cases, metabolic acidosis, hypoglycemia, hypothermia, poor perfusion, shock, and even cardiac arrest may occur [2]. Furthermore, SVT in neonates is relatively rare; therefore, the initial manifestation is often heart failure [4]. A review of the literature revealed three reports of six infants total who exhibited cardiogenic shock from prolonged SVT with normal HR and sinus rhythm on admission [4-6]. Similar to other reports, our patient had normal HR and sinus rhythm initially, and delta waves were not detected in the first ECG [7,8]. Therefore, arrhythmia should be considered, and both ECG and vital signs monitoring should be conducted in critically ill patients. In addition, serial 12-lead ECG is necessary for all patients with arrhythmia.
Early diagnosis is crucial for WPW syndrome because this can lower morbidity and mortality. In neonates, arrhythmia is considered benign. However, if the HR is consistently >180 bpm at rest, there may be hemodynamic instability; hence, ECG should be performed immediately [1]. ECG is the gold standard for the diagnosis of WPW syndrome. WPW syndrome can be diagnosed by a short PR interval (<0.12 seconds), prolonged QRS duration (>0.08 seconds), and delta wave. ECG should be performed for the detection of SVT. In addition, if adenosine is administered after an SVT episode to suppress atrioventricular node conduction, the possibility of finding ventricular pre-excitation through a delta wave increases [9].
Usually, WPW syndrome occurs sporadically and is due to a disorder during embryonic development. However, 3.4% of cases who had first-degree relatives with pre-excitation syndrome are diagnosed as WPW syndrome, which is a known risk factor of WPW syndrome. The familial inheritance pattern is autosomaldominant and related genetic variations are known [10]. Chromosome 20 abnormalities induce HCM with WPW syndrome, and mutations of the protein kinase AMP-activated non-catalytic subunit gamma 2 (PRKAG2) gene on chromosome 7 induces cardiac glycogen overload [11]. Genetic analysis is necessary for understanding the inheritance patterns of familial WPW syndrome and future gene therapy.
Congenital or acquired heart disease is present in 20% of children with WPW syndrome. Up to 37% of infants diagnosed with WPW syndrome may have congenital heart malformations, such as Ebstein anomaly, congenitally corrected transposition of the great arteries, and ventricular septal defects. In 18%, the patients with cardiac rhabdomyomas associated with tuberous sclerosis are diagnosed as WPW syndrome1). Congenital heart disease can cause severe hypotension or cardiac arrest during SVT. Echocardiography is limited in the diagnosis of WPW syndrome; however, it provides information about associated heart disease while providing a view of the structure and function of the heart.
Children with SVT diagnosed in the neonatal period are advised to discontinue the drug by 12 months of age. Gilljam et al. [12] reported that most neonates received antiarrhythmic drug treatment for 6 to 12 months and did not have SVT recurrences. By contrast, SVT associated with ventricular pre-excitation is more likely to persist and requires long-term antiarrhythmic treatment as shown by Riggs et al. [13]. Therefore, in patients in whom arrhythmia is initially difficult to treat and/or those who have ventricular pre-excitation during sinus rhythm, continuation of prophylactic drugs for a longer period was previously considered.
The MYL2 gene encodes the regulator light chain associated with cardiac myosin beta heavy chain. Calcium triggers phosphorylation of the regulatory light chain, which in turn, triggers contraction. Mutations in this gene are associated with conduction defects and mid-left ventricular chamber-type HCM [14]. Andersen et al. [15] reported that more than 50 genes have been associated with HCM, and clinical manifestations can range from asymptomatic to severe disease at an early age. Claes et al. [16] reported that patients who were MYL2 mutation carriers showed a high likelihood of HCM related to hypertension. Missense mutations are the main mutation type found in the MYL2 gene. In diagnostic testing datasets, the c.238A>G (p.Thr80Ala) mutation of the MYL2 gene is a novel variant that has not been reported in the normal population databases (gnomAD, Korean Reference Genome DB [KRGDB]). In silico prediction (SIFT, PolyPhen-2, MutationTaster) predicted that the mutation is deleterious. Moreover, the p.Thr80Ala mutation detected in the present case has an identical amino acid codon position as the p.Thr80Asn mutation, which is a known HCM-related pathogenic mutation [14]. This can serve as further evidence suggesting pathogenicity even if amino acid changes are not the same. Ho et al. [17] reported that a 5-year follow-up of asymptomatic patients with a p.Thr80Asn mutation showed the presence of altered cardiac dimensions in all patients. In our case, no structural abnormality was found on echocardiography. To determine the development of abnormal findings, regular and routine follow-ups are indicated along with the medical evaluation of family members.
In conclusion, this case shows that a neonate with WPW syndrome, presenting with cardiogenic shock and mimicking inborn error of metabolism, can be successfully diagnosed and treated with beta-blockers after suspecting arrhythmia and performing subsequent ECG. Cardiogenic shock should be considered, despite being a rare cause of shock among neonates. Moreover, to determine whether the c.238A>G (p.Thr80Ala) mutation of the MYL2 gene is a pathogenic mutation related to HCM and conduction defects, long-term follow-up and studies are required.
Go to :
 XML Download
XML Download