

 PDF
PDF Citation
Citation Print
Print



서론
색소실조증(incontinentia pigmenti)은 반성 우성(X-linked dominant)으로 유전되는 매우 희귀한 신경 피부 질환으로 출생 시 발병률은 약 100,000명당 1.2명으로 알려져 있다[1]. 이전에 NEMO로 알려진 IKBKG/NEMO (inhibitor of kappa light polypeptide gene enhancer in B-cells, kinase gamma/nuclear factor kappa B essential modulator) 유전자는 성염색체 Xq28에 위치하며 이 유전자의 돌연변이가 남아에서 발병할 경우 매우 치명적이고 대부분 자궁 내에서 사망하므로 이환된 환아는 주로 여아이다[2]. 생존한 색소실조증 환아는 4단계로 구분되는 특징적인 피부 병변을 보이며, 외배엽과 중배엽에서 유래된 중추신경계, 안구, 치아, 모발, 손톱, 유선 등에서 다양한 임상증상을 나타낸다[3]. 특히 출생 시, 혹은 생후 첫 2주 내에 홍반 수포성 병변으로 나타나는 피부 병변은 신생아 선천성 헤르페스 감염, 수두, 수포성 농가진과 같은 감염성 피부 질환의 증상과 혼동될 수 있기 때문에 정확한 감별이 중요하다[4]. 본 저자는 출생 시부터 홍반 수포성 피부 병변을 보인 남아를 감염성 질환으로 오인하였고, 여러 차례 재입원 후 색소실조증으로 진단하였기에 오인했던 경험을 보고하는 바이다.
증례
재태기간 38주 4일, 출생체중 3,090 g (25-50백분위수), 남자 환아로 본원 분만실에서 질식분만으로 태어났다. 산전 초음파상 이상 소견 없었던 환아로 Apgar 점수는 1분 9점, 5분 10점이었고 조기 양막 파수의 증거는 없었으며 분만 시 특이 소견 없었다. 출생 당시 왼쪽 허벅지, 다리에 다수의 수포성 병변이 관찰되어 패혈증 및 피부 감염성 질환에 대한 검사 및 치료를 하기 위하여 본원 신생아중환자실에 입원하였다. 산모 나이 32세로 특별한 기저질환 및 가족력은 없었고, 임신 중 약물 복용이나 감염력은 없었다. 산전 검사상 항문에서 채취한 검체에서 Group B streptococcus 양성으로 분만 4시간 전 항생제를 투여하였다. 이번 출산 이전에 한 번의 유산 이외에 분만력은 없었다. 본원 신생아중환자실 입원 시 시행한 신체검사에서는 체중 3,090 g (25-50백분위수), 신장 49 cm (50백분위수), 두위 35 cm (75-90백분위수)로 측정되었고, 활력징후는 체온 36.8℃, 맥박 150회/분, 호흡 50회/분이었다. 청진상 폐음과 심음에 이상 소견 없었고, 복부 촉진상 간, 비장 비대도 관찰되지 않았다. 피부 소견상 왼쪽 겨드랑이와 옆구리에 갈색의 색소침착이 관찰되었고, 왼쪽 허벅지와 다리에 선상의 수포성 병변 및 갈색 색소 침착이 관찰되었다.
첫 번째 입원 당시 시행한 혈액 검사상 백혈구 13,300/mm3 (호중구 68.9%, 림프구 19.3%, 단핵구 5.6%, 호산구 3.9%), 혈색소 17.3g/dL, 적혈구 용적률 50.2%, 혈소판 168,000/mm3 이었고 C-반응단백질 0.01 mg/dL로 혈액검사상 감염을 시사하는 소견은 없었다. 혈액에서 시행한 toxoplasmosis, rubella, cytomegalovirus, hepes simplex (TORCH) immunoglobulin M (IgM) 검사는 모두 음성이었다. 피부 병변의 배양 검사, 혈액, 소변 배양 검사를 시행하였으며, 피부병변의 감염 질환 감별을 위하여 피부과 협진을 시행하여 mupirocin 성분의 항생제 연고를 도포하며 경과 관찰 하기로 한 후 생후 3일째 퇴원하였다.
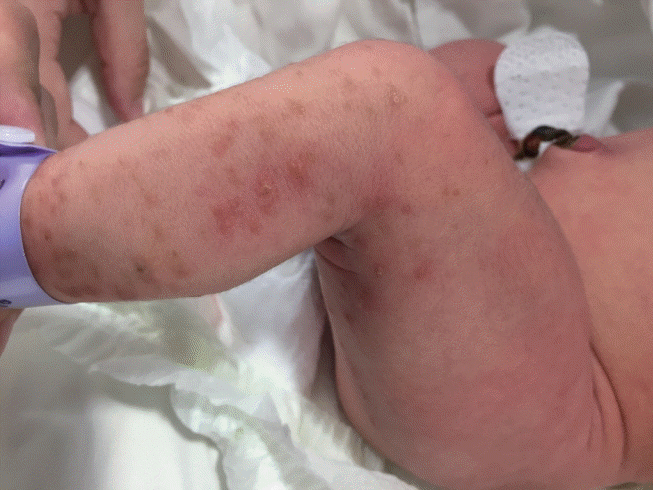
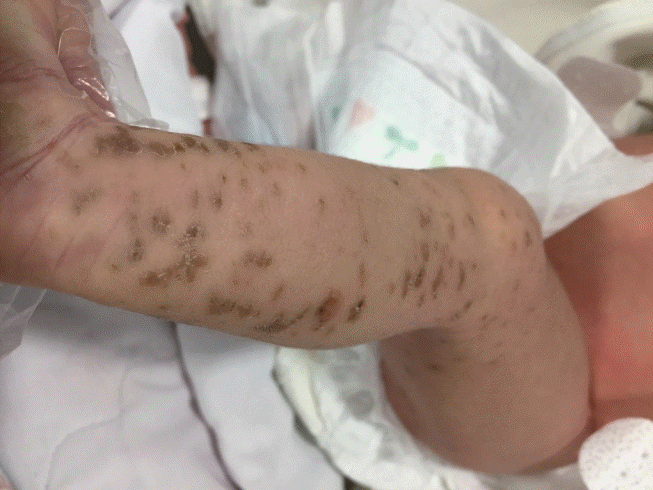
퇴원 다음 날 환아의 왼쪽 겨드랑이와 사타구니, 왼쪽 허벅지에 홍반성 발진 및 수포성 병변이 추가로 발생하여 본원 신생아중환자실에 재입원하였다(Figure 1). 두 번째 재입원 시 시행한 혈액 검사상 백혈구 13,800/mm3, C-반응단백질 0.06 mg/dL로 감염을 시사하는 소견은 없었고, 혈액, 소변 배양 검사는 음성이었으나, 첫 입원 시 시행한 피부 병변 배양 검사상 methicillin-resistant coagulase negative staphylococcus 양성으로 확인되어 vancomycin 정주 투여를 시작하였다. 피부과 재협진을 시행하여 임상적으로 색소실조증의 감별이 필요하지만, 남아이기 때문에 선상표피모반이나 선상 태선을 우선적으로 생각하고, 조직검사는 환아의 나이를 고려하여 보류하기로 하고 mupirocin 성분의 항생제 연고와 국소 스테로이드제 도포를 시행하였다. Vancomycin 5일 정주 후 피부 병변은 갈색의 색소 침착 이외의 수포 병변은 호전되고 추가 병변이 발생하지 않아 생후 11일째 퇴원하였다(Figure 2).
퇴원 다음 날 다시 왼쪽 겨드랑이와 왼쪽 종아리에 홍반 및 수포성 병변이 재발하여 생후 13일째 본원 신생아중환자실에 재입원하였다. 세 번째 입원 시 시행한 혈액 검사에서도 백혈구 14,900/mm3 C-반응단백질 0.02 mg/dL로 감염 소견은 없었으며 herpes simplex virus IgG, varicella zoster virus IgM 모두 음성이었으나, 치료 불충분으로 판단하여 vancomycin 정주 투여를 재개하였다. 피부과 재협진 후 피부 병변에 질산은 습윤 드레싱을 시행하였다. Vancomycin 7일간 정주 투여 후 피부 병변은 호전되어 생후 21일째 퇴원하였다.
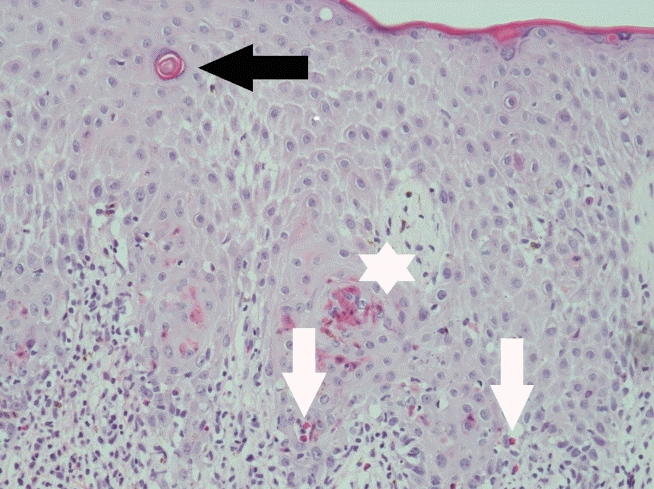
환아는 퇴원 후 왼쪽 겨드랑이와 왼쪽 옆구리, 왼쪽 허벅지와 다리에 수포성 병변이 재발하여 생후 23일째 본원 신생아중환자실에 재입원하였다. 네 번째 입원 시 시행한 혈액 검사상 백혈구 15,700/mm3 C-반응단백질 0.02 mg/dL로 감염 소견은 없었다. 피부 조직 검사를 시행하기로 하였고, 왼쪽 하지의 수포성 병변에서 시행한 조직 검사 결과 표피와 진피에 호산구가 포함된 수포와 해면화가 관찰되고, 진피에서 단핵구의 침윤, 세포의 이상각화증과 keratin pearl의 형성 등 색소실조증에서 관찰할 수 있는 소견들을 확인하였다(Figure 3). 확진을 위하여 유전자 검사를 시행하였고, 말초 혈액을 채취하여 시행한 검사상 정상 남성 핵형인 46, XY를 관찰하였다. 유전자 수준에서의 이상 유무나 염색체의 구조적 이상을 확인하기 위한 분자생물학적 검사는 보호자가 원하지 않아 시행하지 않았다. 색소실조증에 동반될 수 있는 합병증의 유무를 확인하기 위해 뇌 초음파, 청력검사, 안과 협진을 계획하였다. 환아는 입원 기간 동안 신경학적 증상은 보이지 않았으며, 뇌 초음파에서는 특이 소견 관찰되지 않았다. 이비인후과에서 시행한 청력 검사에서 양측 모두 정상 청력을 확인하였다. 안과 검사에서도 사시나 백내장, 망막 박리 등의 이상 소견은 관찰되지 않았고, 생후 27일째 퇴원하였다.
환아는 현재 생후 32개월로 퇴원 후 타 병원에서 외래 추적 관찰중으로, 피부 병변은 생후 60일 이후 호전된 상태이다. 정상 발달 소견 보이고 있어 뇌 자기공명영상검사 등의 추가 검사는 시행하지 않았으며, 치아 발육 또한 결손 및 변형 없이 정상 소견을 보이고 있다.
고찰
색소실조증은 Garrod에 의해 1906년 처음 기술되었고, 1926년 Bloch, 1928년 Sulzberger에 의해 처음 명명되었기 때문에 Bloch-Sulzberger syndrome으로도 불린다[5]. 색소실조증의 출생 시 발병률은 약 100,000명당 1.2명으로 알려져 있고 매년 27.6건의 새로운 케이스가 보고되고 있다. 케이스의 65%-75%는 산발적 돌연변이에 의해 발생하며, 그 외에는 유전성 원인에 해당한다[6].
색소실조증은 매우 드문 신경 외배엽성 질환으로 이전에 NEMO로 알려진 IKBKG/NEMO 유전자의 돌연변이에 의해 발생하는 성염색체 우성 유전 질환이다. X염색체의 장완(Xq28)에 위치하며, 약 80%의 환자에서 4-10번 엑손의 결실이 관찰된다. IKBKG는 nuclear factor-κB (NF-κB) 활성 경로에 필수 단백질인 NEMO/IκB kinase gamma (IKKγ) 를 암호화하는 유전자로서 면역, 염증 반응, 세포 증식, 세포 부착, 외배엽 발생 등 다양한 생리적 기능에 관여한다[7]. IKBKG/NEMO유전자에 돌연변이가 발생하면 NF-κB의 활성이 감소하여, tumor necrosis factor에 의한 세포 자멸에 감수성이 더 증가하여 이로 인해 광범위한 세포 자멸 반응이 나타난다[8]. 남아의 배아는 반접합성으로 이러한 광범위한 세포 자멸 반응이 조기 태아사망의 원인이 되지만, 여아의 배아에서는 X염색체의 비활성화로 인한 기능적 모자이크 반응으로 생존이 가능하다. 때문에 색소실조증의 여아 발생률은 남아에 비하여 월등히 높으며 그 비율은 37:1로 알려져 있다[9]. 남아에서는 클라인펠터 증후군과 같이 여분의 X염색체가 존재하거나, 체성 모자이크 반응, hypomorphic mutation에 의해서 드물지만 생존이 가능하다[10].
색소실조증의 진단기준은 1993년 Landy 와 Donnai [11]에 의해 처음 제안되었고, 2014년 Minic 등[12]이 유전학적 검사를 포함하는 개정된 진단 기준을 발표하였다. 이후 2018년 Rosser [13]에 의해 특징적인 임상증상이 보완된 진단 기준을 제시하였다(Table 1).
색소실조증은 외배엽과 중배엽에서 유래된 여러 기관에서 다양한 임상증상을 보인다. 중요한 진단 기준이 되는 특징적인 피부 병변 외에도 중추신경계, 안구, 치아, 모발, 손톱, 유선 등에 영향을 미친다. 대부분의 색소실조증 환아에서 발견되는 피부 병변은 출생 시, 혹은 생후 첫 2주 내 발생하여 수년에 걸쳐 나타나며 일반적으로 4단계로 나뉜다[11]. 1단계는 약 90%의 환아에서 상, 하지, 두피, 몸통에 홍반성 수포성 병변으로 출생 시, 혹은 생후 첫 2주 내에 발생하여 4개월까지 점진적으로 소실된다. 2단계는 약 70%의 환아에서 과색소 침착된 농포나 가피가 생후 2주에서 6주에 발생하여 6개월까지 소실된다. 3단계는 약 98%의 환아에서 갈색의 색소 침착된 반점이 피부가 접히는 부위와 몸통에 주로 나타난다. 생후 12주에서 26주에 나타나고 병변은 성인이 될 때까지도 나타날 수 있다. 4단계는 약 40% 미만에서 창백함, 위축성 단계로 나타나며 10대 초반에서 성인기에 걸쳐 점진적으로 나타난다. 각 단계의 발생 시기나 지속 기간은 환자에 따라 차이가 있을 수 있으며, 다른 단계의 피부 병변이 동시에 나타나거나 또는 특정 단계가 나타나지 않을 수 있다.
색소실조증은 피부 병변의 단계에 따라 다른 질환을 감별해야 한다. 1단계의 홍반성 수포성 병변은 선천성 헤르페스 감염, 수두, 수포성 표피박리증, 수포성 유천포창과 같은 질환의 증상과 혼동될 수 있다. 2단계의 사마귀 모양의 과각화 단계는 선상 표피 모반, 선상태선, 3단계의 색소 침착 단계에서는 선상 모반 멜라닌 과다증, 4단계의 저색소 침착기에서는 백반증과 같은 멜라닌 저하증과 혼동될 수 있기 때문에 감별이 필요하다[14].
색소실조증 용어는 위와 같은 특징적인 피부 병변의 조직학적 소견에 따른 것인데, 기저 상피층의 멜라닌세포에서 유래된 멜라닌이 진피의 상부에 축적되는 3단계 과색소 침착 단계의 특징에 따라 명명되었다. 본 증례의 환아에서 시행한 조직 검사상 표피와 진피에 호산구가 포함된 수포와 해면화가 관찰되고, 진피에서 단핵구의 침윤, 세포의 이상각화증과 keratin pearl의 형성 등 색소실조증에서 관찰할 수 있는 소견들을 확인 할 수 있었다.
피부 외 증상은 환아의 약 70%-80%에서 관찰된다[15]. 안구에서는 시신경 위축, 사시, 백내장, 포도막염, 망막 박리 등이 나타날 수 있고, 치아에서는 무치증, 발육 부전이 나타날 수 있다. 탈모, 눈썹, 속눈썹의 발생 이상이 나타날 수 있고, 조갑 이상, 부유두 발생이 동반될 수 있다. 약 30%의 색소실조증 환아에서 중추신경계 이상이 발생할 수 있으며, 단순 경련 증상부터 소두증, 뇌경색, 경직성 마비, 정신 지체, 심각한 운동성 장애까지 다양한 증상이 발생할 수 있다.
중추신경계 이상이 동반될 경우 심각한 후유증을 남길 수 있기 때문에 색소실조증의 전형적인 피부 병변이 관찰될 시에는 신경학적 증상이 관찰되지 않더라도 가능한 한 빠른 시일 내에 뇌 자기공명영상검사를 시행하여 중추신경계의 병변 유무를 확인하고, 발달 지연의 가능성에 대한 진단 및 상담이 필요하다[16]. 2013년 Kim 등[17]은 색소실조증의 전형적인 피부 병변을 보이고 신경학적 증상은 관찰되지 않은 생후 4일 여아에서 시행한 뇌 자기공명영상검사에서 다발성 급성 뇌경색 소견을 확인하여 근 긴장도 증가와 성장 발달 지연이 발생하기 전부터 미리 집중적인 추적 관찰을 시행 할 수 있었다. 또한 초기 평가 시에 나타나지 않았던 치아, 안구, 신경학적인 증상이 소아청소년기 이후에도 발현될 수 있기 때문에 타과적 추적 관찰이 지속되어야 한다[13,15].
국내에서 최초로 보고된 남아 색소실조증 증례는 1982년 Park과 Kook [14]에 의해 사마귀양 구진과 수포성 피부 병변과 함께 안진, 백내장 등의 안구 이상 소견이 동반된 증례였다. 이후 1998년 Cho 등[18]에 의해 피부 병변과 안구 및 신경계 이상을 동반한 증례가 발표되었고, 2006년 Kim 등[19]은 수포성 피부 병변과 함께 출생 후부터 3개월까지 간헐적인 간대성 경련의 신경학적 증상이 동반된 남아 색소실조증 사례를 보고하였다. 2006년 Kim 등[15]이 발표한 논문에서는 1995년부터 2005년까지 총 40명의 색소실조증 환아 중 단 2명 만이 남아였다. 2008년 Song 등[20]은 유전자 검사상 정상 핵형을 보인 색소실조증 환아의 증례를 보고하였다.
해외에서는 Hadj-Rabia 등[21]이 2003년 발표한 논문에서 1986년부터 1999년까지 프랑스 내 단일 의료기관의 색소실조증 환자 40명에 대한 증례 발표에서 3명만이 남아였다. 다른 증상 없이 피부 병변만 보인 경우, 피부 병변과 함께 편측 마비의 신경 증상이 동반된 경우, 피부 병변과 함께 안진, 경련의 신경학적 증상, 자기공명영상검사상 전두엽의 허혈성 병변이 관찰된 경우였다. 2006년 Pacheco 등[22]은 9명의 색소실조증 남아의 유전자 검사상 모두 정상 핵형을 보였으나, 1명의 환아가 생후 7세 때에야 경련의 신경학적 증상과 함께 치아의 부식, 벗겨짐의 증상이 나타났음을 보고하였다. 2014년 Fusco 등[23]은 2000년부터 2013년까지 386명의 색소실조증 증례를 조사하였는데 이 중 37명이 남아인 것으로 나타났다. 2019년 Jaramillo 등[16]은 2004년부터 2018년까지 스페인의 두 의료 기관에서 색소실조증으로 진단받은 13명의 환아 모두 여아였음을 보고한 바 있다.
본 증례의 환아는 출생 시부터 몸통과 하지에 수포성 병변이 보였으나 남아이기 때문에 색소실조증보다는 신생아 감염성 질환으로 오인하여 수차례 재입원을 해야 했고 진단이 지연되었다. 신생아의 수포성 피부 질환에 대하여 비감염성 질환, 남아에서의 색소실조증의 발병 가능성을 염두하여 불필요한 광범위 항생제 치료를 하지 않도록 해야 할 것이다.
 XML Download
XML Download