

 PDF
PDF Citation
Citation Print
Print



INTRODUCTION
Baller-Gerold syndrome (BGS) is a rare genetic disorder, also referred to as craniosynostosis-radial aplasia syndrome, which was first described independently by Baller [1] in 1950 and Gerold [2] in 1959. It is characterized by craniosynostosis, particularly of the coronal sutures, a dysmorphic face, such as a prominent forehead, ocular proptosis, hypertelorism, and a small mouth, and limb abnormalities, especially in the upper extremities, including missing fingers and malformed or absent thumbs [3,4]. In addition to phenotypic variability, additional features presented in BGS include failure to thrive, developmental delay, and systemic anomalies such as congenital heart disease (ventricular septal defect and patent ductus arteriosus), renal agenesis, an imperforate anus, and nervous system abnormalities (agenesis of the corpus callosum, or seizure disorders) [5]. The pattern of inheritance in BGS is thought to be autosomal recessive, and the condition is mainly caused by mutations in the RecQ like helicase 4 (RECQL4) gene located on chromosome 8q24.3, which encodes RecQ helicases [6]. Here, I report the first Korean case of a premature infant diagnosed as BGS with craniosynostosis of the coronal sutures, thumb hypoplasia of both hands, and a mutation in the RECQL4 gene confirmed by diagnostic whole exome sequencing (WES).
CASE REPORT
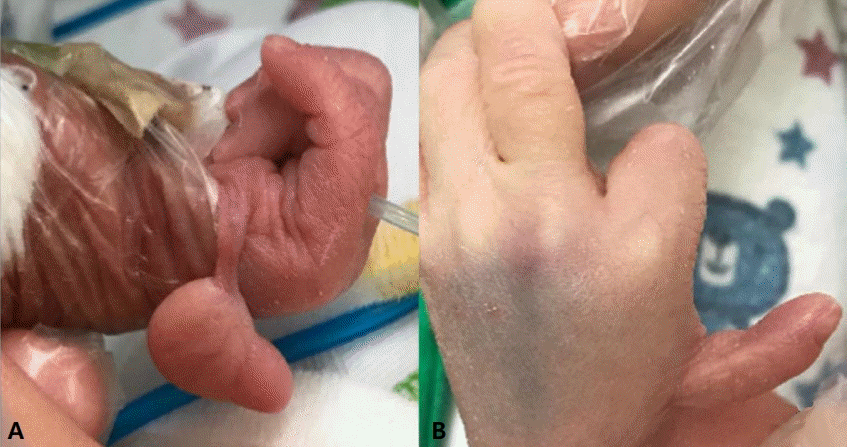
A 1-day-old female infant weighing 1,490 g was born at 35 weeks and 1 day of gestational age by repeat cesarean section because of preterm prelabor rupture of the membrane and unconrolled preterm labor. Apgar scores were 7 at 1 minute and 9 at 5 minutes. The pregnancy and delivery were uneventful despite suspected right thumb anomaly on the prenatal ultrasonography. There was no exposure of teratogenic agents, including sodium valproate during pregnancy. A family history of genetic disorders or metabolic diseases was unknown. At birth, her body weight was 1,490 g (<10th percentile), which was defined as small for the gestational age, height was 37 cm (<10th percentile), and head circumference was 28.5 cm (<10th percentile). Vital signs were as follows: blood pressure, 61/34 mm Hg; heart rate, 141 beats/minute; respiratory rate, 68 beats/minute; and body temperature, 36.5℃. Physical examination on admission revealed a palpable ridge on the bilateral coronal sutures, a dysmorphic face, such as a prominent forehead, a high arched palate, and low set ears, a floating thumb on the right hand, and a malformed thumb with a tight web space between the thumb and index finger on the left hand (Figure 1). The limited movement of both elbows, which were not fully extended (130°) was also observed.
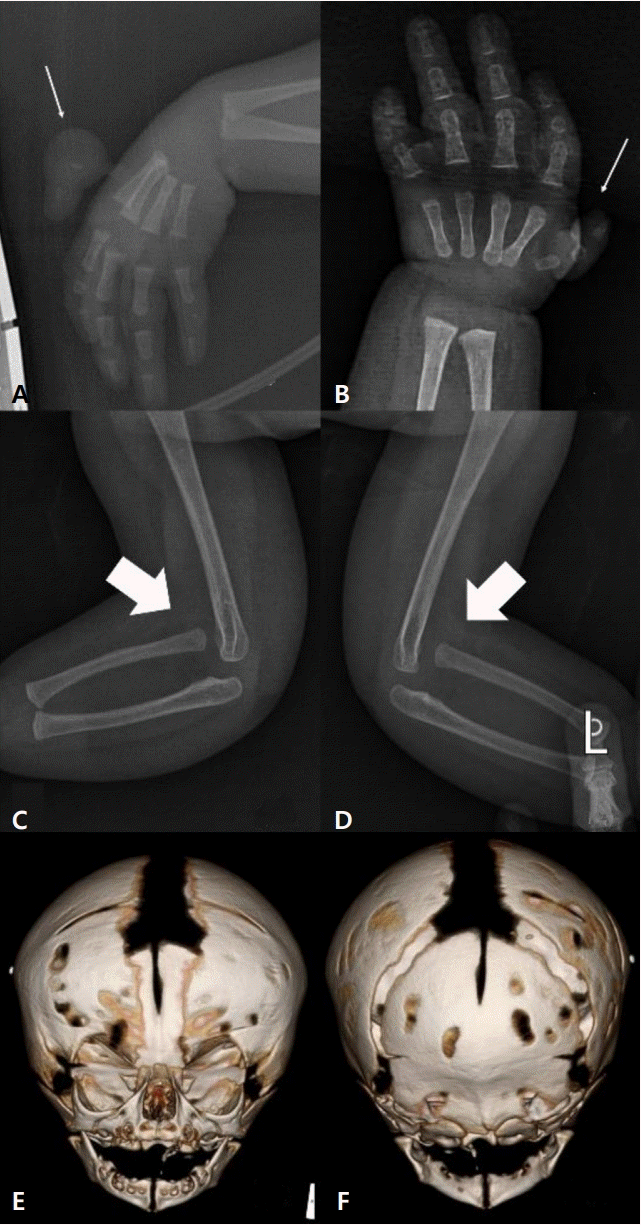
Initial blood test results were non-specific. The initial infantogram showed a mild diffuse haziness with interstitial opacities, air bronchograms on both lung fields, and the dislocated joint of both elbows. A few rudimentary bones in both hypoplastic thumbs were detected by a hand X-ray. On the 24th day after birth, serial skull X-rays revealed the early fusion of the coronal sutures. On the 27th day, we confirmed craniosynostosis of the bilateral coronal sutures by three-dimensional (3D) head computed tomography (CT) (Figure 2). Brain ultrasonography, magnetic resonance imaging, echocardiography, and abdominal ultrasonography were performed to evaluate associated anomalies and the results of these were normal.
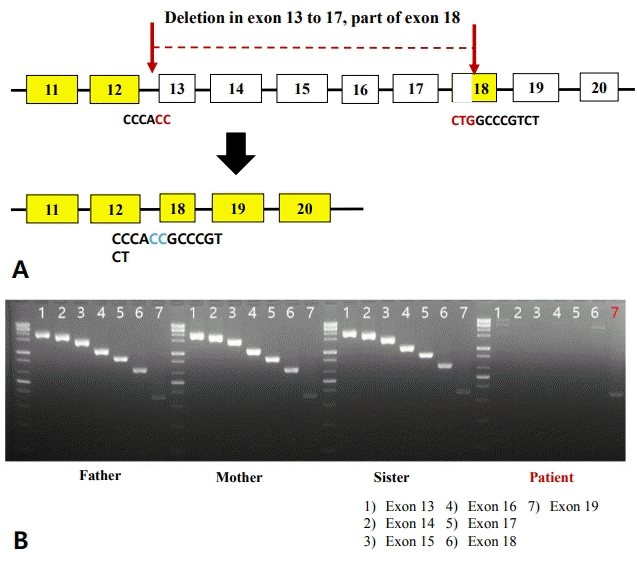
Genetic testing was conducted to evaluate the cause of multiple anomalies. Peripheral chromosome analysis showed a karyotype of 46, XX, and chromosomal microarray results were normal. To identify genetic disorders, diagnostic WES was performed and a homozygous deletion from exon 13 to 19 in the RECQL4 gene was detected, which was consistent with BGS associated with craniosynostosis and upper limb anomalies at birth. Direct polymerase chain reaction (PCR) and sequencing on the RECQL4 gene (exon 13 to 19) in the patient confirmed a homozygous deletion from exon 13 to 17 and in part of exon 18 in the RECQL4 gene (Figure 3). Direct PCR and sequencing on the RECQL4 gene in the parents and sister were performed for genetic counseling and the results were normal suggesting that the deletion was de novo.
The patient received continuous positive airway pressure (CPAP) therapy because of progressive respiratory distress, including moaning sounds, mild chest retraction, and low oxygen saturation (80% to 90%) at 1 hour after birth, which was diagnosed as mild respiratory distress syndrome (RDS) based on the findings of the initial chest X-ray. On the 5th day after birth, she had no limitation of movement in both elbows, which were fully extended, and the infantogram showed the spontaneous reduction of her dislocated elbow joints. Following the improvement in RDS, she was weaned from CPAP on the 5th day and achieved full enteral feeding on the 9th day after admission. At 37 weeks of corrected gestational age, her clinical condition was stable with normal growth velocity and she was weaned from the incubator. She was discharged from the neonatal intensive care unit at 41 days after birth without oxygen supplementation and the need for a gastric nutrition tube.
The patient was followed up to monitor growth and developmental status, craniosynostosis, and thumb hypoplasia in the outpatient neonatal, pediatric neurologic, neurosurgery, and orthopedic clinics after discharge. On the 125th day of life, she underwent an operation for the removal of the right hypoplastic thumb with necrotic change. At that time, the growth velocity of the head circumference was low (less than 1 cm per month). In addition, there were palpable ridges on the sagittal and lambdoid sutures on physical examination and progressive premature fusion of all the cranial sutures was confirmed by a follow-up 3D-head CT. At 6 months of corrected gestational age (222 days after birth), she underwent cranioplasty, which consisted of craniotomy and distractor fixation. Currently, the patient is undergoing bone distraction to achieve the desired head shape with regular follow-up to monitor growth and development. At the follow-up at 6 months of corrected gestational age, the patient had not grown up to catch-up, her weight (5.7 kg), height (60.0 cm), and head circumference (39.9 cm) were still <10th percentile for the corrected gestational age. However, her development was appropriate for the corrected gestational age. Pollicization of the left hypoplastic thumb would be performed at around 12 months of life.
DISCUSSION
BGS is a rare, autosomal recessive disorder. The exact prevalence of BGS, which affects approximately less than 1 per million people, is unknown, and fewer than 40 cases have been reported in the medical literature [7]. Based on previous reports, the key features of BGS include early fusion of the cranial sutures, especially coronal sutures, and pre-axial limb malformation (radius or thumbs), which are apparent at birth [8]. Therefore, BGS is a genetic disorder that should be suspected in neonates with craniosynostosis and upper limb malformation at birth, such as in this case, where premature fusion of the coronal sutures and hypoplastic thumbs were observed.
Craniosynostosis involving any or all sutures is a core feature that can be used to differentiate BGS from other syndromes with upper limb malformation and hematopoietic abnormalities, such as thrombocytopenia-absent radius syndrome and Holt-Oram syndrome [5,9]. Although the involvement of the coronal sutures (alone or in combination) is most common (64%), all cranial sutures may be affected, such as the metopic (36%), lambdoid (27%), and sagittal (9%) sutures [8]. The clinical severity of craniosynostosis in BGS is variable; plagiocephaly, a prominent forehead, and ocular proptosis may be presented with premature fusion of the cranial sutures. In this case, craniosynostosis of the coronal sutures was immediately detected after birth and progressive premature fusion of all cranial sutures were observed at 3 months of life, which resulted in microcephaly, a prominent forehead, bulging eyes, and shallow orbits.
The defect of pre-axial upper extremities is another major feature of BGS, which has been observed in previous patients. The patient in this case had hypoplastic thumbs on both hands (type II for the left thumb and type IV for the right thumb). The absence or hypoplasia of thumbs is the most common deformity in BGS; it occurs in 95% of patients, followed by the absence or hypoplasia of radii in 77% of patients. The defects of upper extremities (excluding the pre-axial portion) include anomalies of the phalanges of fingers and metacarpal bones in 95% of cases, a short or curved ulnar in 82% of cases, and fused or absent carpal bones in 27% of cases [5]. Various skeletal anomalies have been reported, such as vertebral defects, including scoliosis, missing vertebrae, flat vertebral bodies, and abnormal toes (absent or hypoplastic phalanges of toes). Multiple malformations associated with visceral organs have also been reported in the literature. The visceral defects observed in BGS patients include polymicrogyria, hydrocephalus, corpus callosum agenesis in the central nervous system, ventricular septal defects, tetralogy of Fallot, subaortic valvular hypertrophy in the cardiovascular system, kidney ectopia, hydronephrosis in the urogenital system, and an imperforated anus [9-11].
Mutations in the DNA helicase of the RECQL4 gene have been identified mainly in patients with BGS [6]. Kaneko et al. [4] reported a homozygous deletion from intron 12 to the former part of exon 18 in the RECQL4 gene in three patients with BGS, which was similar to the mutation of the RECQL4 gene detected in my patient with homozygous deletion from exon 13 to 18 in the RECQL4 gene. Mutations in the RECQL4 gene have been associated with two other autosomal recessive disorders: Rothmund-Thomson syndrome (RTS) and RAPADILINO syndrome [12,13]. Therefore, RTS and RAPADILINO syndrome should be excluded in the diagnosis of BGS based on the distinctive clinical presentation of each disorder. RTS is characterized by poikiloderma, sparse hair in the eyelashes and eyebrows, small stature, skeletal and dental abnormalities, juvenile cataracts, and increased risk of cancers such as osteosarcoma and skin cancer [12]. RAPADILINO syndrome is characterized by radial ray defects, patellae hypoplasia or aplasia, a cleft or highly arched palate, diarrhea, dislocated joints, a small size, limb malformation, a slender nose, and normal intelligence [14]. To differentiate BGS from RTS and RAPADILINO syndrome, major symptoms or symptoms essential for the diagnosis of each disorder should be considered. Although the presence of craniosynostosis and radial hypoplasia occurring simultaneously at birth may be a diagnostic criterion for BGS, patients diagnosed as RTS or RAPADILINO syndrome have also been reported to present phenotypes overlapping with those of BGS [13]. Further studies are needed to define distinctive features for the diagnosis of BGS, RTS, and RAPADILINO syndrome and to determine whether the three RECQL4-associated diseases may be regarded as separate disorders or belonging to the same clinical spectrum.
The treatment for the symptoms of BGS depends on the clinical manifestation. Craniosynostosis may require surgical treatment such as craniotomy, which is determined by neurosurgery or craniofacial specialists. Surgery for bilateral craniosynostosis is recommended before 6 months of age. Our patient underwent craniotomy and distractor fixation at 6 months of corrected age because of the progression of fusion of the coronal sutures. In BGS patients with absent or hypoplastic thumbs, pollicization of the index finger is an appropriate method to restore functional grasp; however, many patients with aplastic or hypoplastic thumbs can grasp objects without orthopedic surgery [15]. The human DNA helicase, RECQL4, is known to play an important role in DNA replication, transcription, recombination, and repair. Recent biochemical and molecular studies found that mutations in the RECQL4 gene could cause genomic instability and carcinogenesis [16]. Debeljak et al. [17] reported a case where BGS was presented with lymphoma. In addition, pathogenic RECQL4 mutations in patients with RTS may be associated with an increased risk for developing osteosarcoma [18]. Therefore, BGS patients with pathogenic RECQL4 mutations, such as in this case, should be closely observed for symptoms or signs of malignancies such as osteosarcoma and lymphoma.
In conclusion, BGS should be considered in infants with an early fusion of a specific cranial suture and malformation of the upper limb extremities at birth. To differentiate from other genetic disorders with overlapping features, careful physical examination to detect key symptoms or signs for the diagnosis of BGS is necessary and a proper genetic test that includes the RECQL4 gene is needed to confirm BGS. This case report describes a premature infant with BGS presenting with early fusion of the coronal sutures and hypoplastic thumbs at birth and harboring a mutation in the RECQL4 gene. To the best of my knowledge, this is the first case of RECQL4-related BGS in Korea.
 XML Download
XML Download