

 PDF
PDF Citation
Citation Print
Print



INTRODUCTION
Congenital pulmonary airway malformation (CPAM) is a developmental anomaly of the lungs that occurs in 1 in 10,000 to 35,000 newborn infants. It originates from the bronchopulmonary foregut and is caused by delayed pulmonary development at 4 to 7 weeks of gestation [1]. CPAM accounts for 95% of congenital cystic pulmonary lesions, most commonly found via prenatal screening at 18 to 20 weeks of gestation [2]. Predicting the presence and severity of respiratory symptoms in patients with prenatally diagnosed CPAM is difficult, because the use of prenatal ultrasound diagnosis and the role of other associated factors in the presence and onset of symptoms are unclear [3]. As patients with CPAM can possibly undergo malignant transformation and are predisposed to repetitive infection, surgical treatment in the early stages has been the current treatment of choice. However, there is an on-going controversy regarding elective surgical resection in infants with asymptomatic CPAM [4].
In this study, we report our experience with the clinical and imaging features as well as the postnatal management of newborns with prenatally diagnosed CPAM at a single tertiary neonatal center over 12 years. By comparing the reliability of imaging tools and identifying potential predictive factors associated with respiratory symptoms at birth and during infancy, we provide comprehensive clinical data that can lead to the proper management of CPAM.
MATERIALS AND METHODS
We conducted a retrospective analysis of the medical records of 66 patients with prenatally diagnosed CPAM delivered at Severance Children’s Hospital between January 2005 and July 2017. The histopathological types of CPAM have traditionally been based on the Stocker classification [5]. For newborns with prenatally diagnosed CPAM, postnatal chest radiography, lung ultrasonography, and/or chest computed tomography (CT) were performed. The patients’ respiratory symptoms at birth and during infancy, history of surgery, surgical methods and outcomes, and postoperative pathological findings were reviewed. We also analyzed the long-term follow-up data of patients with CPAM at the department of pediatrics and thoracic surgery.
The study protocol was approved by the Institutional Review Board of Severance Hospital, Yonsei University College of Medicine, Seoul, Korea (4-2019-0137). The requirement for informed consent was waived because of the retrospective nature of the study and because the analysis used anonymous clinical data. Statistical analysis was performed using IBM SPSS Statistics for Windows/Macintosh, version 23.0 (IBM Corp., Armonk, NY, USA). Noncontinuous variables between the two groups were compared using the chi-square test or Fisher’s exact test, and the outcomes were represented as percentages. Continuous variables between the two groups were compared using the two-sample T-test or Mann-Whitney U-test, and the outcomes were represented as mean±standard deviations. All outcomes with P-values <0.05 were considered statistically significant.
RESULTS
1. Characteristics of newborn infants and mothers
The mean gestational age at the time of delivery was 38.8 weeks (38.8±1.4), and the mean birth weight was 3,050 g (3,050±460). The ratio of boys to girls was 1:1, with 33 patients of each sex. The mean final follow-up duration was 48.4 months (48.4± 27.5) (Table 1). The mean Apgar scores for 1 and 5 minutes were 6.4 and 8.0, respectively. The mean age of the mothers was 30.3±4.0 years. Analysis of the obstetric history revealed only one case of accompanying polyhydramnios. The timing of prenatal diagnosis of CPAM was at a gestational age of 22.6 weeks (22.6±3.1).
2. Postnatal diagnostic examinations
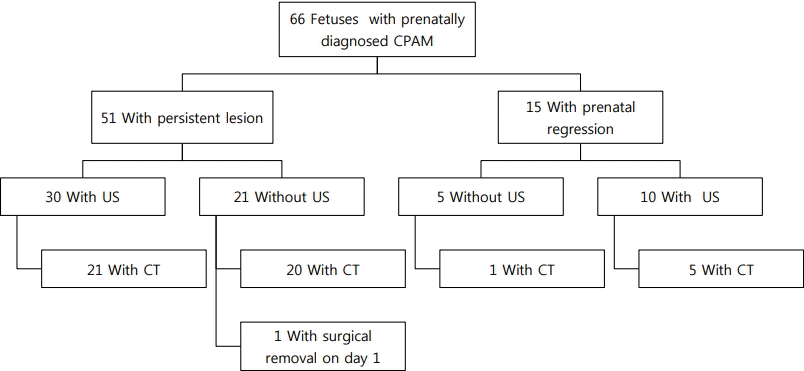
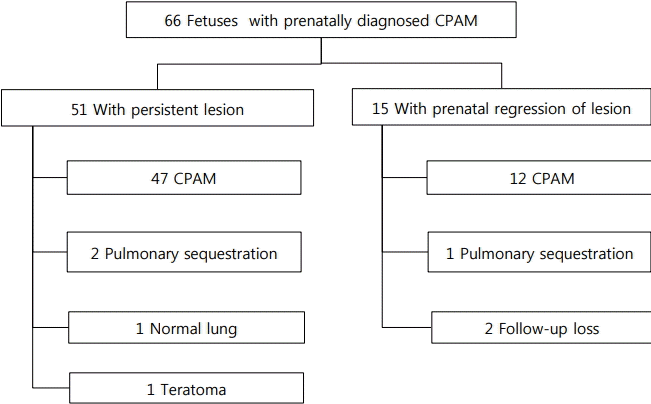
Chest radiography was performed in all patients at birth. Lung ultrasonography, chest CT, or both examinations were performed at the request of a neonatologist and thoracic surgeon. Among patients with prenatally diagnosed CPAM, 15 (23%) showed prenatal regression of the lung lesions throughout the serial follow-up via prenatal ultrasonography (Figure 1). However, 12 patients with CPAM and one with pulmonary sequestration were diagnosed via postnatal imaging studies (Figure 2). One of these patients had no abnormal chest radiography and lung ultrasonography findings after birth and did not exhibit notable clinical symptoms during infancy. However, pulmonary sequestration was diagnosed on the basis of routine chest CT performed at the age of 1 year. Two patients were lost to follow-up.
Fifty-one patients with prenatally diagnosed CPAM exhibited persistent lung lesions until birth. Lung ultrasonography was performed in 30 patients. In 21 of these patients, chest CT was also performed (Figure 1). Twenty patients who did not undergo lung ultrasonography underwent chest CT, except for one patient who underwent emergency operation on the day of birth. Among the 51 patients, 47 were diagnosed with CPAM, two with pulmonary sequestration, and one with mature cystic teratoma (Figure 2). However, one patient showed normal findings, as confirmed by lung ultrasonography and chest CT. The patient with mature cystic teratoma was found to have a mediastinal mass, mimicking CPAM, on prenatal ultrasonography. Chest radiography performed after birth revealed soft-tissue opacity in the lower mediastinum, and ultrasonography demonstrated a large lobulated cystic mass at the posterior mediastinum and within the peritoneum. Chest CT revealed a lobulated cystic mass at the posterior mediastinum and upper abdomen extending through the diaphragmatic hiatus. Laparoscopic surgery was performed, and the pathology was confirmed as mature cystic teratoma.
Collectively, among 66 patients with prenatally diagnosed CPAM, 59 had CPAM, three had pulmonary sequestration, one had cystic teratoma, and one had normal findings, as confirmed postnatally. Twenty-two patients (33%) had no visible lung lesions on postnatal chest radiography. Of these 22 patients, nine underwent lung ultrasonography; among them, five had normal ultrasonography findings, but were later diagnosed with masslike lung lesions via chest CT. The remaining 44 patients showed varying types of lung lesions on chest radiography performed after birth: 21 showed opacity, 13 had cystic lesions, seven showed hyperinflation, and three had pneumothorax. Although 13 patients showed normal findings on lung ultrasonography, six were later diagnosed with mass-like lung lesions via chest CT. The frequencies of lung lesions on the left and right sides were identical in patients with CPAM. Chest radiography and/or lung ultrasonography were not adequately reliable for diagnosing CPAM because chest CT identified lung lesions in patients with prenatally regressed CPAM as well as postnatally negative chest radiography or lung ultrasonography findings. The sensitivity and positive predictive value of lung ultrasonography, chest radiography, and chest CT in the diagnosis of CPAM were 60% and 83%, 70% and 88%, and 100% and 90%, respectively (Table 2). Therefore, chest CT was the most reliable imaging technique for diagnosing CPAM.
3. Accompanying malformations
In the entire patient cohort, 12 had cardiac malformations, seven had kidney malformations, and one had diaphragmatic hernia (Table 1). Cardiac malformations included atrial septal defect, patent ductus arteriosus, ventricular septal defect, and coarctation of the aorta. Kidney malformations included pelviectasis and hydronephrosis.
4. Respiratory symptoms at birth and during infancy
We sought to identify the factors associated with clinical symptoms, including the occurrence of respiratory symptoms, at birth and during infancy. Twenty-five patients (38%) exhibited respiratory symptoms at birth, and among them, three who exhibited pneumothorax required chest tube insertion and 11 required invasive ventilator support. The remaining 41 patients (62%) were asymptomatic at birth. Respiratory symptoms included tachypnea, gasping that required oxygen therapy, intercostal retraction, and symptoms that required invasive treatment, such as chest tube insertion or intubation. Sex, gestational age, birth weight, associated malformations, histologic types of CPAM, perinatal regression of CPAM, and location of lung lesion of the patient, as well as the age of the mother, were not significantly associated with the presence of respiratory symptoms at birth (Table 3). The maximum size of the lesion reached during pregnancy and timing of diagnosis were also not associated with the presence of respiratory symptoms at birth. The Apgar scores at 1 and 5 minutes (P=0.006 and P=0.012, respectively) and mediastinal shift on chest radiography (P =0.023) were significantly correlated with the respiratory symptoms at birth (Table 3).
Of the 12 patients (17%) with respiratory symptoms during infancy, 10 were admitted to the hospital because of pneumonia or bronchiolitis. Respiratory symptoms during infancy included cough with fever, wheezing, and pneumonia confirmed via chest radiography. The mean age at admission was 2.8±2.2 months. No significant prenatal and postnatal characteristics could predict the presence of respiratory symptoms during infancy.
5. Surgery and complications
Among the 59 patients with CPAM confirmed after birth, 21 (35%) underwent surgery, and the mean age at surgery was 21 months (ranging from birth to 54 months). No significant difference was observed in patient characteristics between the surgery and nonsurgery groups (Table 4). However, in the surgery group, the proportion of patients who presented respiratory symptoms at birth was higher (48%) than that in the nonsurgery group (34%), albeit not statistically significant.
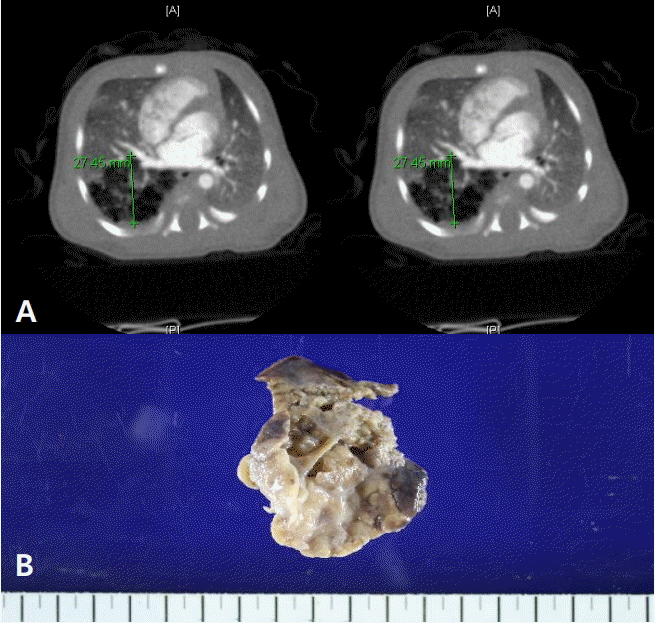
Among the 25 patients presenting respiratory symptoms at birth, three underwent early surgery during the neonatal period. One patient underwent right lung segmentectomy at 6 days after birth because of pneumothorax. Another patient underwent right lower lobectomy via video-assisted thoracoscopy (VATS) at 21 days after birth because of right pneumothorax. Chest CT revealed multicystic masses filled with air (Figure 3A), and the surgical and pathological findings were consistent with type 2 CPAM (Figure 3B).
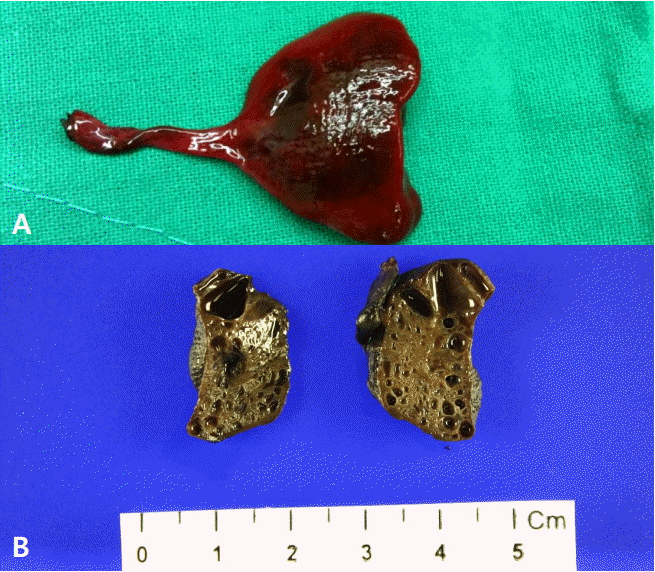
Another patient, exhibiting right congenital diaphragmatic hernia combined with CPAM, underwent bilateral lobectomy via thoracotomy and repair of congenital diaphragmatic hernia on the day of birth. However, a day after surgery, the patient succumbed to severe respiratory failure. This patient’s pathological findings were consistent with type 1 CPAM (Figure 4). Meanwhile, one patient had a huge, multi-segmented, inoperable cystic lesion with an air-fluid level in the right thorax, and underwent cyst thoracentesis 4 days after birth. Twenty-two days after the procedure, the patient succumbed to severe respiratory failure, despite chest tube insertion and ventilator care. A pathological examination was not performed.
Most of the surgeries were lobectomies performed via VATS (15 patients, 71%). Three patients had postoperative complications, including postoperative fever, chylothorax, and pleural effusion requiring thoracentesis. Two patients were readmitted because of postoperative pneumonia. Postoperative histological examinations confirmed the diagnosis of CPAM. According to the Stocker classification (Table 5), nine patients had type 1, 11 had type 2, and one had type 3 CPAM. Accompanying inflammation was observed in three patients.
The mean follow-up duration of the 21 patients who underwent surgery was 46.6 months. Among the 21 patients, four were hospitalized, and the mean age at hospitalization was 2.3 months. Most of these patients were admitted because of pneumonia within 6 months of age, and one patient was hospitalized because of mycoplasma pneumonia at the age of 35 months.
DISCUSSION
Herein, we have reported our experience with CPAM over 12 years at a tertiary center. A strength of this study is that we compared imaging tools and attempted to identify various potential predictive factors associated with the occurrence of respiratory symptoms at birth and during infancy, which are essential for postnatal management. Since the diagnosis and management of CPAM remain controversial, by reviewing many patients over a long term, we aimed to provide comprehensive clinical data on CPAM that could lead to its proper management.
CPAM generally occurs in the unilateral form, but exhibits higher incidence on the right side [6]. It may be accompanied by excessive amniotic fluid, nonimmune fetal hydrops, ascites, and mediastinal shift, as well as other structural anomalies. However, CPAM is not associated with chromosomal abnormalities [7]. In our study, the frequency of CPAM was identical between the left and right lungs. Polyhydramnios was observed; however, it was not significantly correlated with the clinical outcome. No patients had chromosomal abnormalities. The histopathological types of CPAM have traditionally been based on the Stocker classification [5]. This classification was based on the histological and morphological findings of lung lesions obtained via surgery. The original classification initially included three types of CPAM (types 1, 2, and 3), and types 0 and 4 were added later [5,8]. Type 1 is the most common type. However, in this study, type 2 accounted for the highest proportion (11/21, 52.4%), and no patient had type 0 or 4.
The diagnosis of CPAM requires a multidisciplinary approach involving obstetricians, radiologists, pediatricians, surgeons, and pathologists [9]. As ultrasonography technology has developed, the sensitivity of both prenatal and postnatal diagnosis of CPAM has been increasing [10]. One study reported that the prenatal diagnosis rate of CPAM was as high as 85.7% over 8 years of retrospective assessment [11]. Although the sensitivity and specificity of prenatal magnetic resonance imaging is as high as 95%, prenatal ultrasonography is the most commonly used diagnostic method [12]. Lesions may grow until 25 to 28 weeks of gestation, but may partially regress during the third trimester of pregnancy [13]. However, the regression of lesions observed under prenatal ultrasonography may not result in complete loss of the lesion [14]. In this study, although 15 patients had regression of their lesions, 13 of them displayed lesions after birth. Thus, even if the lesion regresses during prenatal screening, postnatal imaging is essential for the accurate diagnosis of CPAM. In a previous study assessing 26 patients with prenatally diagnosed CPAM, chest CT was highly sensitive and showed a good positive predictive value that was consistent with our results [15]. Prenatal ultrasonography, postnatal chest radiography, or lung ultrasonography alone is not adequately reliable to diagnose CPAM or its complete regression [16]. We suggest chest CT for confirming the diagnosis of CPAM, considering its high sensitivity and positive predictive value.
Respiratory symptoms observed immediately after birth in patients with prenatally diagnosed CPAM are extremely diverse, and may necessitate mechanical ventilation or immediate surgical intervention. Identifying the risk factors for respiratory symptoms allows the medical staff to provide detailed and precise consultation to the caregivers, as well as adequate treatment to the patients as early as possible. Moreover, such infants should preferably be delivered at medical centers with neonatal intensive care units. A previous study suggested that a high CPAM to volume ratio (CVR), excessive amniotic fluid, and the presence of ascites are good prognostic factors for respiratory symptoms [14]. More specifically, CVR ≥1.6 was reported to increase the risk of fetal hydrops [17]. Much attention has been drawn to identifying prognostic factors predicting respiratory outcomes, such as contralateral lung volume per thoracic volume and the incidence of mediastinal shift [18]. However, many studies failed to identify the prognostic factors predicting the respiratory symptoms in patients with CPAM [19]. In this study, multiple factors were analyzed to identify the prognostic factors for respiratory symptoms at birth and during infancy. CVR was excluded because it was not routinely measured in all examinations. Instead, we compared the maximum size of the lesion by measuring its largest dimension reached during pregnancy. In our study, the Apgar scores at 1 and 5 minutes and mediastinal shift on chest radiography were significantly associated with respiratory symptoms at birth. Other factors were found not to be significant.
Different opinions exist regarding the treatment of CPAM. For prenatal treatment, thoracoamniotic shunt or multiple courses of antenatal betamethasone for high-risk fetal CPAM are considered effective [20,21]. In our study, no patients had received prenatal intervention. Moreover, the standard for postnatal treatment has not yet been established. If the patient is symptomatic, surgical treatment is recommended [20]. The symptoms include infection, pulmonary hemorrhage, pneumothorax, respiratory difficulty, and malignant transformation. Pneumonectomy is the most commonly used surgical treatment for patients with CPAM, and it has a high success rate [11,22]. A previous study that assessed 1,120 infants demonstrated that the thoracoscopic approach induced fewer complications and resulted in shorter hospitalization [23]. In neonates, thoracoscopic intervention may be difficult because of the limited operative space within their small bodies [24]. However, minimal-access surgery is becoming widespread, and recently published series have reported increased utilization of VATS from 32.2% in 2008 to 48.2% in 2014 [23]. In our study, 15 patients (68%) underwent VATS, with a high success rate; therefore, we confirm that thoracoscopic surgery can be performed both safely and effectively.
There is an on-going debate about surgical interventions performed in asymptomatic newborn patients. The risk of CPAM lesions becoming symptomatic in the first 5 years is less than 5% [4]. However, even for asymptomatic patients, the possibility of repeated infection and malignant transformation are the reasons for pneumonectomy, which may outweigh the benefits of conservative management. CPAM can occasionally be a cystic malformation, and it may be a pleuropulmonary blastoma induced by DICER1 mutation, albeit rarely (with an estimated incidence of 1 in 300,000) [20,25]. During follow-up, as infants and children are inevitably and repeatedly exposed to radiation from radiological studies, the European Academic Society suggests that only physical examination is required for lesions that are asymptomatic and stable until the patient becomes an adult [26]. However, there are concerns suggesting that in the absence of surgical resection, patients and families may experience continuous and unnecessary stress throughout the long-term follow-up process [20]. A previous study reported that approximately 25% of asymptomatic patients with CPAM eventually develop symptoms at an average age of 6 to 7 months, which is different from our findings (2.8±2.2 months) [19]. Our study was performed at a tertiary center to which patients with high-risk CPAM are referred and where a high proportion of respiratory symptoms are seen at birth; these factors could have accounted for the shorter average onset time of symptoms observed in our study than in other studies. Very few previous studies performed long-term follow-ups in pediatric patients who underwent early surgical treatment. At our center, the mean follow-up duration of the 21 patients who underwent surgery was 46.6 months.
A limitation of this study is that it was performed retrospectively, and different obstetric specialists were involved in the prenatal ultrasound examination. Furthermore, although our center aimed to apply an identical treatment protocol for all patients, different thoracic surgeons were involved in the treatment of patients and the treatment period varied according to the severity of the patients’ symptoms. Despite these limitations, this study provides meaningful experiences because patients with prenatally diagnosed CPAM underwent long-term follow-ups and were treated at a single tertiary center.
Overall, this study provides comprehensive clinical data on prenatal and postnatal clinical characteristics, diagnostic imaging, respiratory symptoms, treatment, and long-term follow-up of infants with prenatally diagnosed CPAM at a single tertiary center. Prenatal and postnatal ultrasonography or chest radiography alone provided unreliable diagnostic accuracy. They showed lower sensitivity and positive predictive value in the diagnosis of CPAM than did chest CT (with a sensitivity of 100% and positive predictive value of 90%). In case of unclear or inconsistent findings on ultrasonography or radiography, chest CT can be a valuable diagnostic tool for confirmation. Low Apgar scores and mediastinal shift on radiography can be predictive factors for respiratory symptoms at birth. However, symptoms during infancy were not associated with prenatal and postnatal factors. Therefore, chest CT combined with periodic monitoring of clinical symptoms is important for the proper diagnosis and management of patients with prenatally diagnosed CPAM and to guide appropriate timing of surgery.
 XML Download
XML Download