

 PDF
PDF Citation
Citation Print
Print



INTODUCTION
Congenital pancreatic cysts are a rare condition [1]. Sometimes, pancreatic pseudocysts are caused by trauma to the pancreas or pancreatitis. A true congenital pancreatic cyst is extremely rare in newborn babies [2]. This type of cyst can be caused by developmental abnormalities of the pancreatic duct, and the resulting sequestration of the primitive pancreatic duct [1-4]. Prenatal diagnosis is difficult due to their extreme rarity. Therefore, the diagnosis of a congenital pancreatic cyst can be delayed until a few months after birth, when the infant presents with an abdominal mass, abdominal distension, or vomiting due to a gastric outlet obstruction. Excision of the cyst is the treatment of choice [3].
Multiple congenital pancreatic cysts may be accompanied by systemic diseases or other congenital conditions such as von Hippel-Lindau syndrome, polycystic kidney disease, Ivemark syndrome, trisomy 13, trisomy 15, and Meckel-Gruber syndrome [5]. Cystic fibrosis can also lead to cystic dilatation of pancreatic ducts [5]. A congenital pancreatic cyst should be considered if the fetus has an abdominal cyst without a definite origin. Early diagnosis with evaluation of the accompanying disease is required. A prompt diagnosis is crucial to prevent fatal complications such as cholangitis, pancreatitis, cyst rupture, and peritonitis [6]. This case report discusses a newborn infant with multiple pancreatic cysts diagnosed by magnetic resonance cholangio-pancreatography (MRCP). These cysts were suspected prenatally to be stomach diverticulum or duplication cysts of the intestine by fetal ultrasound.
Go to : 
CASE REPORT
This study was approved by the Institutional Review Board of Busan Paik Hospital (19-0030), and a waiver of consent was granted for chart review without patient contact.
A 35-year-old primigravida woman was referred to our medical center for further evaluation of fetal intraabdominal cysts in a twin baby. A prenatal ultrasonography in our medical center suspected these cysts to be stomach diverticulum or duplication cysts of the intestine (Figure 1).
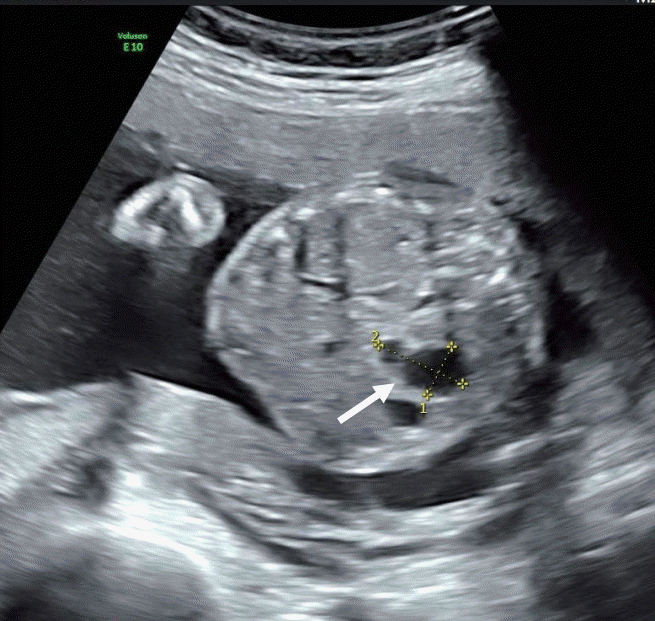
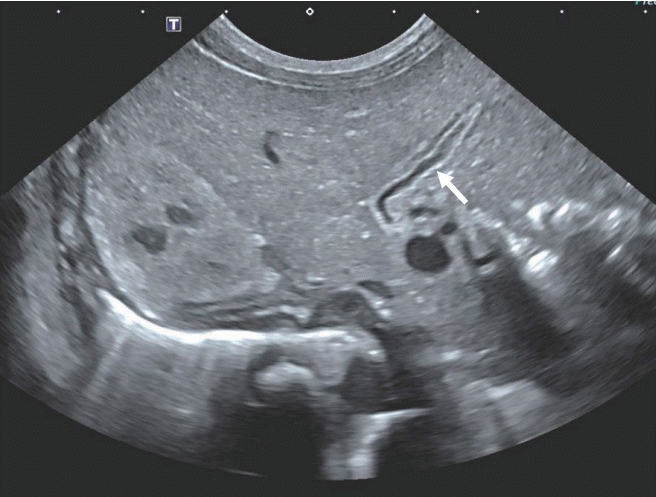
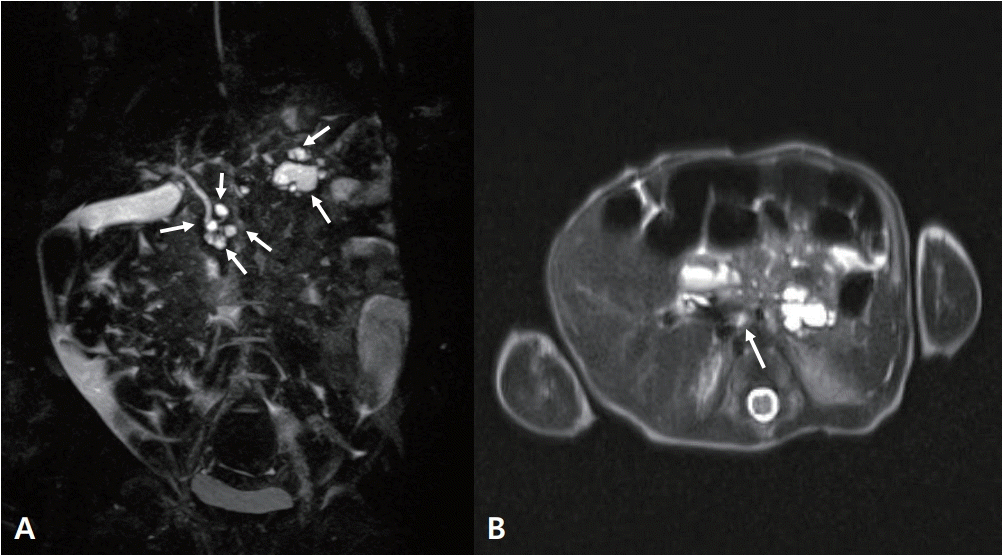
At 36+5 weeks, a cesarean delivery was performed and male twins were born. The weight of the first baby was 2.9 kg (50th percentile). However, the weight of the second baby was 1.9 kg (<3rd percentile). The second baby was much smaller than the first, and thus, he was hospitalized in the neonatal intensive care unit. His height was 45 cm (3rd to 10th percentile) and his head circumference was 31.5 cm (10th to 25th percentile). His Apgar score was 6 at 1 minute and 8 at 5 minutes. Direct bilirubin and γ-glutamyl-transferase (GGT) started to elevate 2 weeks after birth. However, he did not show signs of hemolytic anemia causing direct hyperbilirubinemia. His direct bilirubin level was 2.0 mg/dL, with a total bilirubin level of 4.9 mg/dL, GGT level of 240 U/L (reference, 13 to 147), and alkaline phosphatase (ALP) level of 634 U/L (reference, 145 to 420). His serum glucose level of 70 to 90 mg/dL was normal (reference, 50 to 90). We conducted an abdominal ultrasonography at the 16th postnatal day that showed a collapsed gallbladder, strongly suggestive of congenital biliary atresia (Figure 2). An MRCP was performed to confirm congenital biliary atresia on the 22nd postnatal day; it showed multiple, variable sized cysts in the pancreatic head and tail, with the largest one (16 mm in diameter) located in the pancreatic tail (Figure 3). The levels of amylase and lipase were normal: amylase, 6 IU/L; lipase, 14 U/L.
To investigate associated congenital malformations, further evaluations were performed. The echocardiogram findings were normal, other than the detection of a small atrial septal defect 2.6 mm in size. There were no abnormalities in the cranial, renal, or sacral ultrasonography. A chromosome analysis showed a normal male karyotype.
We started treatment using ursodoxycholic acid, and then direct bilirubin and GGT started to decrease and normalized at discharge from the hospital (direct bil irubin, 0.8 mg/dL; GGT, 57 U/L). The total bilirubin level was 2.7 mg/dL and ALP was 1,029 U/L. As discussed above, his amylase and lipase levels had been normal, and were 11 IU/L and 10 U/L at discharge, respectively.
The baby was discharged on the 34th postnatal day. He was clinically well without jaundice or an acholic stool. Exploratory laparotomy or excision of cysts had not been performed yet. He is still under regular radiologic and laboratory checkups for pancreatic and hepatic enzymes. If either of these findings are aggravated, we will proceed with an operation.
Go to :
DISCUSSION
Congenital pancreatic cysts are extremely rare in newborns; there are only 25 reported cases [1,5]. Pancreatic cysts are divided into six types: congenital-developmental, retention, duplication, pseudocysts, neoplastic, and parasitic [2,7,8]. True pancreatic cysts are lined with true epithelium and include congenital developmental cysts, retention cysts, and duplication cysts [8]. Pseudocysts are the most common cysts of the pancreas. They develop after pancreatic fat necrosis is resorbed due to injury or inflammation of the pancreas. They are surrounded by granulation tissue and a fibrous capsule without an epithelial lining [9]. Neoplastic cysts can be either a mucinous type such as an intraductal papillary mucinous neoplasm, or a serous (clear-cell) type such as serous cystadenoma [9].
Congenital pancreatic cysts are usually asymptomatic. However, they can present as an abdominal mass, abdominal distension, or vomiting due to a gastric outlet obstruction or cyst rupture. They can also present as cholangitis, pancreatitis, or peritonitis [1,5,6,8]. Our patient did not present with any of the clinical symptoms previously reported.
The location of congenital pancreatic cysts are more commonly in the tail or neck of the pancreas (62%), as opposed to the head (32%) [1,2,8]. However, 6% of the cysts are located in the overall pancreas [5]. In this reported case, the multifocal pancreatic cysts were located in both the head and tail of the pancreas.
Sepulveda et al. [10] reviewed eight reported cases of prenatally diagnosed congenital pancreatic cysts and found that the cyst is part of a systemic disease in five (63%) out of eight cases. Congenital cysts may be associated with systemic diseases such as von Hippel-Lindau syndrome, polycystic kidney disease, Beckwith-Wiedemann syndrome, and asphyxiating thoracic dysplasia [5,6,9-11]. Therefore, clinicians should determine whether an associated systemic disease is present with the cyst. Clinicians also need to examine the kidneys, liver, bile duct, ovaries, mesentery, and urethra [11]. In our case, the congenital pancreatic cyst was an isolated finding without other associated systemic diseases.
Radiologic findings and laboratory results are important when diagnosing a congenital pancreatic cyst [7]. Our case was diagnosed using MRCP. The most commonly used radiologic diagnostic tools include the abdominal computed tomography (CT) and abdominal magnetic resonance imaging (MRI) [7]. However, some researchers have used an abdominal ultrasonography, and if there are any suspicious findings, a CT or MRI is also performed to confirm the diagnosis. Further, some researchers have aspirated the cystic fluid in adults, if the diagnosis was not certain based on radiologic findings [12]. If the level of lipase in the cystic fluid is high, then the cyst can be considered to be a pancreatic cyst [7].
Excision of the congenital pancreatic cyst is the treatment of choice. These type of cysts are usually located in the body or tail, allowing for complete excision to be easily performed [3,11]. If the cyst is located in the tail, excision with distal pancreatectomy is the preferred treatment. If the size of the cyst is increasing rapidly with clinical symptoms, prompt surgical intervention is needed [3]. Unlike our case with multifocal cysts located in the pancreatic head and tail, Choi et al. [13] reported that an isolated 56×47 mm homogenous cyst originating from the pancreatic tail was excised with a distal pancreatectomy. However, excision of the cyst is not always the treatment of choice for all patients. Treatment varies according to the location and size of the cyst [5]. Warnock et al. [5] performed excision of multiple pancreatic cysts with distal pancreatectomy, because the baby had severe gastric outlet obstruction symptoms. Conversely, if multifocal cysts are present in the whole pancreas or if small cysts are located in the head, clinicians should consider waiting, with regular follow up with radiologic imaging, rather than performing immediate surgery. Our patient has multifocal cysts in the pancreatic head and tail, and a cystectomy or pancreatectomy has not been performed yet.
In conclusion, a congenital pancreatic cyst is an extremely rare lesion in newborns, and prenatal diagnosis is difficult due to this rarity. A congenital pancreatic cyst should be considered if the fetus has an abdominal cyst without a definite origin. A prompt diagnosis is crucial to prevent fatal complications.
Go to :
 XML Download
XML Download