

 PDF
PDF Citation
Citation Print
Print



Dear Editor,
The most common genomic abnormality in Philadelphia chromosome-negative myeloproliferative neoplasms (MPNs) is a functional V617F mutation in the Janus kinase 2 (JAK2) kinase-like structural domain [1, 2]. Although chromosomal translocation involving JAK2 is rare, it has been reported in hematological malignancies [3]. Because of similar clinical features, experts advocate that all rare JAK2 rearrangement cases should be classified into the same category [4]. We report a case of MPN with BCR-JAK2 and BCR-PPP1R32 rearrangements, showing t(9;22;11)(p24;q11.2;q13). To our knowledge, this is the first report of PPP1R32 as a novel fusion partner of BCR. Written informed consent was obtained from the patient for publication of this case report and accompanying images. This case study was approved by the Institutional Review Board of Army Medical University, Chongqing, China (No.2021-219).
A 56-year-old man was admitted to our outpatient clinic in March 2020 for fatigue and left upper abdominal pain. Peripheral blood examination indicated a white blood cell (WBC) count of 178.1×109/L (25.0% neutrophils, 4.0% lymphocytes, 1.0% monocytes, 4.0% eosinophils, 0% basophils, 18.0% late granulocytes, 15.0% neutrophilic bands, 12.0% promyelocytes, 21.0% myelocytes), an eosinophil count of 7.1×109/L, a platelet count of 162.0×109/L, and 125 g/L Hb. A bone marrow (BM) aspirate indicated abnormal granulocyte proliferation (1.5% myeloblasts, 2.0% promyelocytes, 27.5% myelocytes, 32.0% late granulocytes, 20.0% neutrophilic bands, and 5.5% eosinophils), mainly in middle- and late-stage cells, some of which had large bodies and coarse A particles, suggesting MPN (Fig. 1A-C). BM biopsy showed active granulocyte proliferation, with all stages of late cells being visible, and fibrous tissue hyperplasia, suggesting MPN with primary myelofibrosis or MPN unclassifiable. Flow cytometry indicated an increased granulocyte ratio, suggesting CML. FISH and real-time reverse transcription (RT)-qPCR did not reveal the BCR–ABL1 fusion gene and JAK2 V617F mutation, but FISH showed BCR amplification in 90.0% of cells in interphase (Fig. 1D). Cytogenetic analysis revealed a 46,XY,t(9;22;11)(p24;q11.2;q13) [20] karyotype (Fig. 1E). Therefore, MPN was diagnosed.
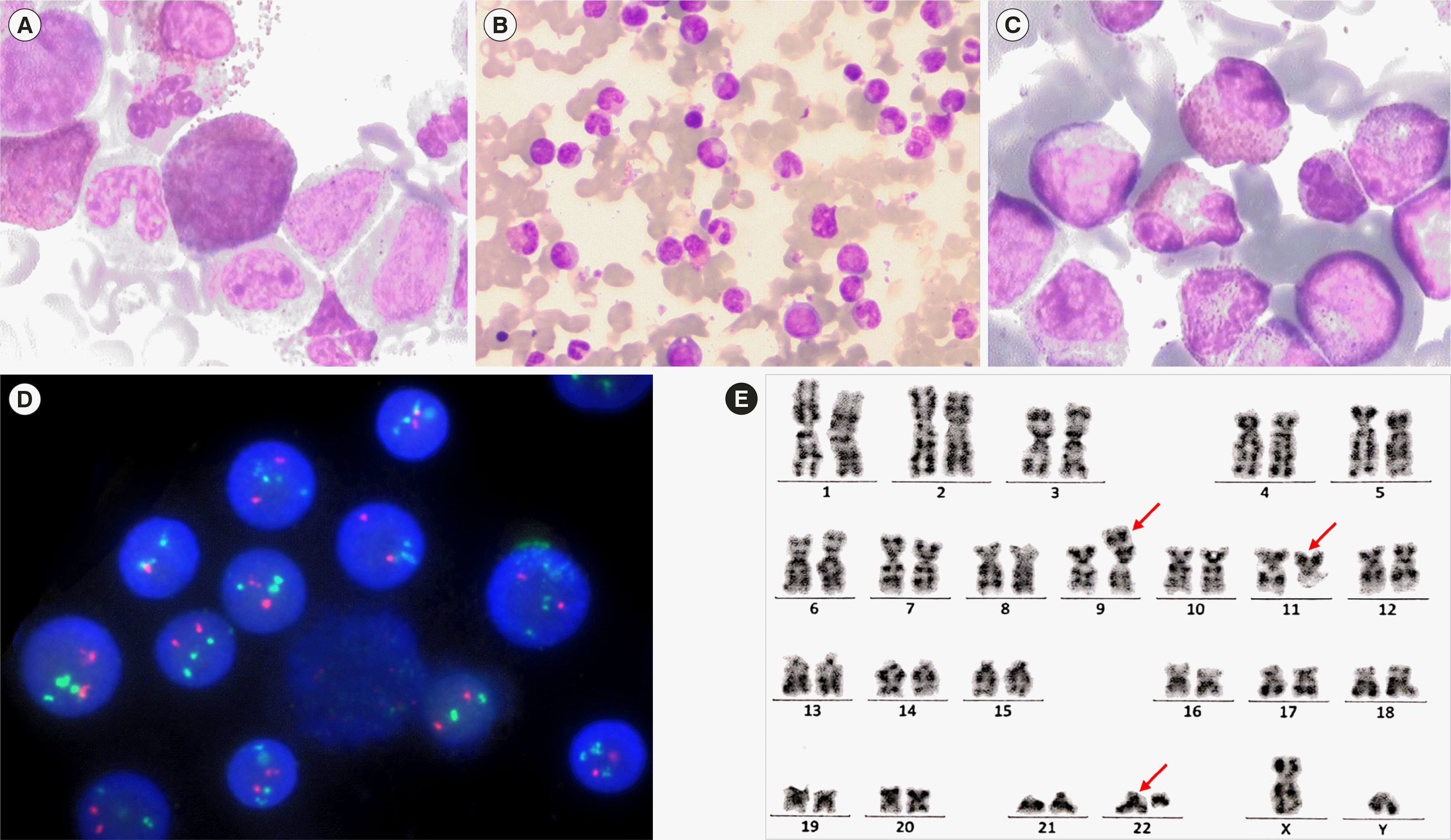 | Fig. 1Bone marrow and peripheral blood smears, BCR–ABL1 FISH, and chromosomal karyotype of this patient. (A) Peripheral blood smear demonstrating leukocytosis with left shift (Wright’s staining, ×400). (B) Bone marrow aspirate smear showing severely depressed proliferation of the erythroid lineage, without dysplastic changes in erythroid cells (Wright’s staining, ×400). (C) Bone marrow aspirate smear showing hyperplastic granulocytic marrow with abnormal granulocyte proliferation (mainly in middle- and late-stage cells) and eosinophilia (Wright’s staining, ×400). (D) BCR–ABL1 FISH analysis of the Philadelphia chromosome does not reveal the BCR–ABL1 fusion gene. However, a BCR probe signal is observed in 90% of cells in interphase (green signal). (E) G-banded chromosome analysis (bone marrow) reveals an abnormal 46,XY,t(9;22;11)(p24;q11.2;q13)[20] karyotype. The red arrows represent the chromosomes where translocation occurs.
|
After treatment with hydroxyurea and interferon for 10 months, the patient’s WBC count decreased to normal. However, the patient discontinued the treatment on his own for three months because of a low platelet count. Peripheral blood examination was repeated in January 2021, revealing a WBC count of 54.9 ×109/L, a platelet count of 118.0×109/L, and 70 g/L Hb. Given that BCR is located on 22q11 and JAK2 on 9p24 and that rearrangement between BCR and JAK2 has been reported in MPN patients [3, 5], we used RT-qPCR to detect the BCR–JAK2 fusion gene, revealing a positive result, with a copy number-to-internal reference gene copy number ratio of 0.00713.
Following PCR, Sanger sequencing on an ABI 3130 Genetic Analyzer (Life Technologies, Carlsbad, CA, USA) identified in-frame fusions of BCR exon 1 to JAK2 exon 17 (Fig. 2A). Using second-generation sequencing of the fusion gene, we identified a BCR–PPP1R32 fusion gene in addition to the BCR–JAK2 fusion gene (Fig. 2B and C). Based on shared features and WHO classification [6], cases in which translocation leads to fusion genes with JAK2 (especially, ETV6–JAK2 and BCR–JAK2) can be considered variants of myeloid/lymphoid neoplasms with PCM1–JAK2. In conclusion, the patient was diagnosed as having MPN with BCR–JAK2/PPP1R32. The patient continued treatment with hydroxyurea and interferon, and after two years of follow-up, he has currently achieved clinical and hematological remission.
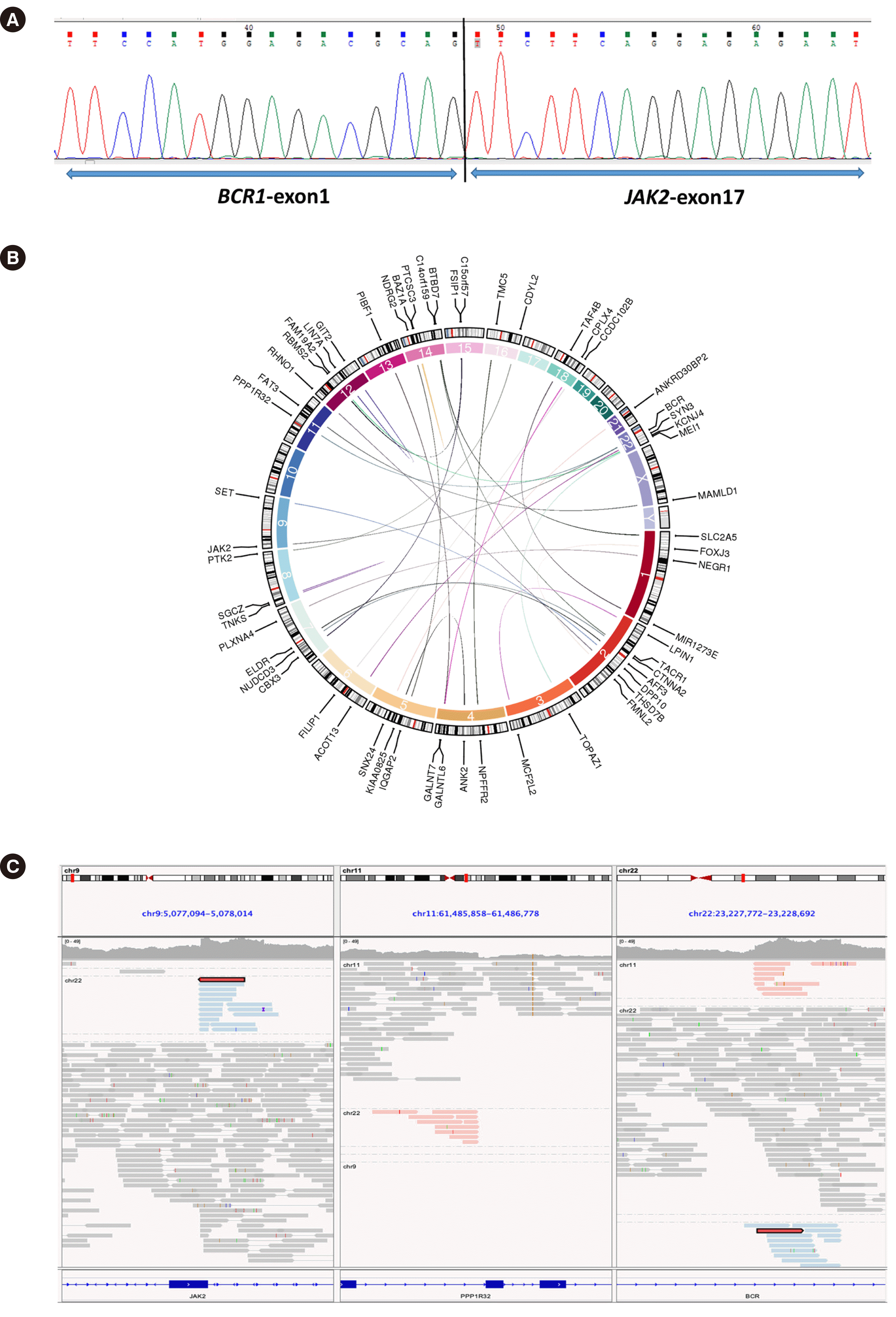 | Fig. 2Typical gene fusions detected by sequencing technology in this patient. (A) Direct sequencing and sequence alignment of reverse transcription-PCR products show that exon 1 of BCR is fused with exon 17 of JAK2. (B) Typical fusion genes, including BCR–JAK2 and BCR–PPP1R32, detected by second-generation sequencing. Lines connect fusion genes, and the circle represents the position of the chromosome where the gene is located. (C) Integrative Genomics Viewer snapshot displaying the BCR–JAK2 and BCR–PPP1R32 fusions.
|
The BCR–JAK2 fusion gene is rare but has been reported in cases of ALL, AML, MPN, and atypical CML, indicating that the BCR–JAK2 rearrangement lacks lineage specificity [3]. The BCR–JAK2 fusion gene triggers Stat5 phosphorylation and the expression of Stat5 target genes, such as the anti-apoptotic Bcl-xL gene, thereby promoting tumorigenic properties and improving cell survival [7]. JAK2 inhibitors may be effective against hematological tumors with BCR–JAK2 fusion genes, but the response may be transient or even ineffective [5]. Therefore, the use of JAK2 inhibitors may be limited to bridging therapy before allogeneic hematopoietic stem cell transplantation.
We identified a novel fusion partner of BCR, PPP1R32, in addition to JAK2. PPP1R32 is located at 11q12.2, which does not exactly match the karyotype position (11q13) in our patient. Given the higher accuracy of second-generation sequencing results of fusion genes, positional differences were considered minor errors in karyotype analysis. PPP1R32 has an unknown function but is indispensable for maintaining adherens junctions of polarized ependymal cells. Changes in PPP1R32 protein levels during ependymal cell differentiation may increase ventricular lesions, including tumor transformation [8]. However, its function in hematologic diseases remains to be investigated. With the advent of molecular testing, JAK2 and BCR rearrangement-related malignancies have garnered public attention. Our understanding of the clinical phenotypes of these rare diseases must improve, and new, targeted drugs must be developed to improve patient survival.
Go to : 
 XML Download
XML Download