

 PDF
PDF Citation
Citation Print
Print



INTRODUCTION
Next-generation sequencing (NGS)-based approaches for clinical evaluation of infants with suspected genetic diseases have expanded substantially in recent years [1-3]. In particular, whole-genome sequencing (WGS) and whole-exome sequencing (WES) have improved the diagnosis of rare genetic diseases in critically ill neonates and infants. Compared with conventional genetic testing, rapid genomic testing (RGT) permits accurate genetic diagnosis of many patients [4-6]. Recent studies have suggested that RGT is faster and more reliable than standard WES and WGS [7-9]. NGS-based RGT enables rapid and timely diagnosis of rare genetic diseases.
Early diagnosis of incurable genetic diseases and profound disability in newborns raises several ethical issues, such as parents making premature decisions as the bonding with the baby is not fully established [10]. Moreover, RGT poses a substantial clinical and laboratory burden [8]. A successful diagnosis implementation requires changing the existing workflow by a multidisciplinary team, including intensive-care therapists, clinical geneticists, and clinical pathologists. Furthermore, the cost of rapid WGS and WES is two to four times that of standard WGS and WES [10]. The cost of analysis of the large amount of data generated by WGS or WES is inevitably higher than that of conventional gene sequencing [11]. Many related findings and variants of uncertain significance (VUSs) increase the risk of inaccurate diagnosis and the turnaround time (TAT) of result interpretation. Therefore, WGS or WES in infants and children with genetic diseases is not a viable option, especially in developing countries.
If actionable genes are targeted, ethical approval can be waived, which reduces the TAT. Applying variant interpretation and pathogenicity prediction models can further reduce the TAT and the risk of inaccurate diagnosis. Therefore, we designed an RGT platform to specifically sequence 254 medically actionable genes associated with preventable or curable Mendelian diseases using dried blood spot (DBS) samples of patients.
MATERIALS AND METHODS
Patient enrollment
Patients were prospectively enrolled based on the following criteria: (1) abnormalities in routine neonatal screening testing (NST); (2) unexplained neonatal hypotonia or neonate-onset seizures without exponential causes such as prematurity, malformations, and perinatal insults; (3) unexplained and abnormal laboratory findings, such as liver enzymes, metabolic acidosis, and hyperammonemia, suggesting hidden metabolic diseases; (4) skeletal dysplasia or joint problems suggestive of storage diseases; and (5) neurodevelopmental delay with abnormal results in metabolic screening testing. This study was approved by the Institutional Review Board of Seoul National University Hospital, Seoul, Korea (No. H-2002-069-1101). Informed consent for genetic testing was obtained from all patients and their guardians in the case of minor patients.
NEOseq_ACTION panel design
The NEOseq_ACTION panel was designed to diagnose genetic diseases in neonates or during early infancy that require a rapid and precise diagnosis to ensure prompt and efficient treatment. The NEOseq_ACTION is a comprehensive NGS panel that includes 254 genes that were selected based on consultations with clinical and metabolic geneticists. The selected genes include potentially actionable genes associated with diseases exposed by neonatal metabolic screening performed in Korea and diseases listed on the US Recommended Uniform Screening Panel Core and Secondary Conditions of Advisory Committee on Heritable Disease in Newborns and Children (https://www.hrsa.gov/advisory-committees/heritable-disorders/rusp/index.html).
The diseases targeted in this panel can be classified as: glycogen storage diseases; adrenal insufficiency; galactosemia; inborn errors of amino acid, organic acid, and fatty acid metabolism; treatable or curable lysosomal storage diseases; immunodeficiency; hemoglobinopathy; genetic hyperbilirubinemia; congenital and genetic sensorineural hearing loss; and other genetic diseases, such as cystic fibrosis, mineral metabolism disorders, and congenital myasthenic syndromes (curable diseases: Table 1; full list: Supplemental Data Table S1).
Retrospective validation of the NEOseq_ACTION panel
The validation set included data from 24 patients with known pathogenic single nucleotide variants (SNVs) and small insertion or deletions (indels) across 22 genes (Supplemental Data Table S2). All validation samples were confirmed through biochemical testing. In total, 99.8% and 98.96% of bases were covered by at least 1- and 20-fold coverage, respectively. The median percentage of on-target reads across samples was 78.2%. The mean depth of coverage was 142×. All 40 variants in the 24 cases were detected using the NEOseq_ACTION panel.
Sample processing, target enrichment, and sequencing
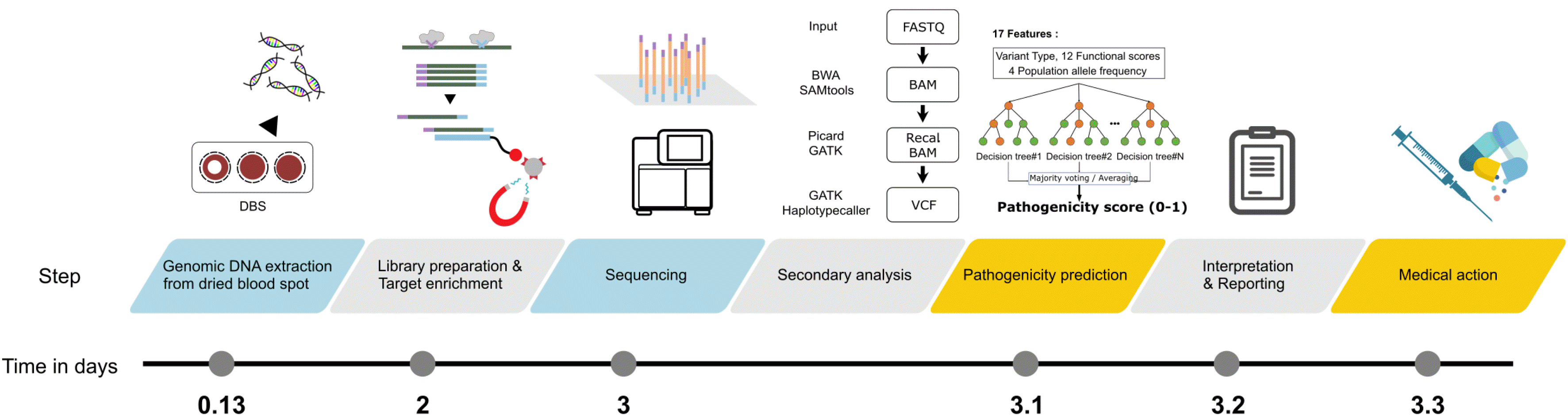
Genomic DNA was extracted from two out of three DBSs (Fig. 1) using a Chemagic 360 instrument (PerkinElmer, Baesweiler, Germany) and fragmented using a Nextera DNA library preparation kit (Illumina, San Diego, CA, USA). Libraries were prepared from 100 ng of total genomic DNA using the Nextera DNA library preparation kit, according to the manufacturer’s instructions. Paired-end 150-bp sequencing was performed on the MiSeqDx platform (Illumina). Raw sequencing data were obtained in the FASTQ format.
Pipeline for variant calling
The sequencing reads were aligned to the human genome reference sequence (GRCh37) using the Burrow–Wheeler Aligner (BWA-MEM, version 0.7.5) [12]. The aligned sequence reads (SAM file) were sorted, indexed, and converted to BAM files using SAMtools (version 1.9) [13]. Duplicate sequences in the BAM files were removed using Picard tools (version 2.18.2) (https://broadinstitute.github.io/picard/). Next, local realignment and base recalibration of the BAM files were performed using the Base Recalibrator in GATK (version 4.0.3.0) [14] based on the dbSNP (version 138), Mills, and 1000G gold standard indel databases. SNVs and indels were identified using the GATK Haplotype Caller tool. Called variants were annotated using SnpEff [15].
Variant interpretation and pathogenicity prediction model
Called variants were interpreted using the MedyCVi software (MedySapiens, Seoul, Korea) based on InterVar [16]—a bioinformatics tool for the interpretation of genetic variants according to the American College of Medical Genetics and Genomics and Association for Molecular Pathology guidelines [17] based on databases of disease-associated genetic variants such as ClinVar [18] and Human Genome Mutation Database (HGMD) [19]. A machine learning-based prediction model was developed to support clinical decisions in variant interpretation based on estimation of the pathogenicity of each VUS.
The MedyPatho (MedySapiens) prediction model predicts the pathogenicity of non-synonymous SNVs based on nine functional scores (PolyPhen [20], MutPred [21], Sorting Intolerant From Tolerant [22], Protein Variant Effect Analyzer [23], Variant Effect Scoring Tool [24], Likelihood Ratio Test [25], MutationAssessor [26], MutationTaster [27], Fathmm [28]), two conservation scores (GERP [29] and PhastCons [30]), and the population allele frequency from four databases (Genome Aggregation Database [31], Exome Aggregation Consortium [32], Korean Reference Genome [33], and Korean Variant Archive [34].
The prediction model was trained and evaluated using a dataset compiled from the variant databases of ClinVar and HGMD. Specifically, only missense variants were selected, and for the ClinVar database, variants with a review status labeled as “criteria provided, multiple submitters, no conflicts; criteria provided, single submitter; or reviewed by expert panel” were considered. In addition, pathogenic variants marked as disease-causing mutation or disease-associated polymorphism in the HGMD were included. The final dataset comprised 143,555 pathogenic and 86,326 benign variants.
The variants added to ClinVar since 2020 were considered as test data. Variants for which protein sequences were similar to those in the training set were excluded using the CD-HIT program [35] based on a sequence identity threshold of 80%. The prediction model was trained with 43,590 pathogenic and 26,499 benign variants and was subsequently evaluated with 8,295 pathogenic and 8,630 and benign variants.
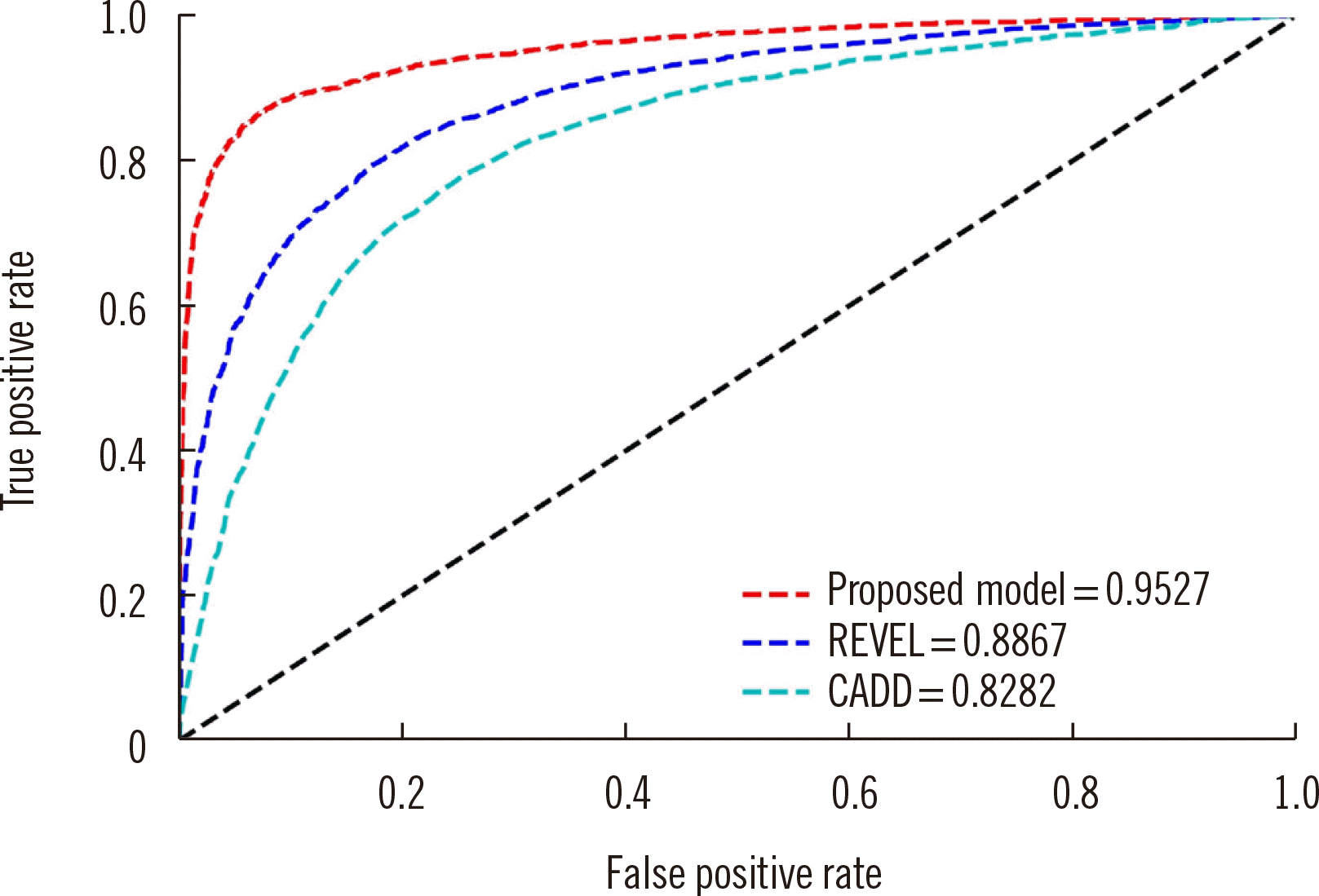
The prediction model was built using LightGBM [36]—a tree-based gradient boosting model. Performance was evaluated in comparison with two in silico prediction tools (Rare Exome Variant Ensemble Learner [REVEL] [37] and Combined Annotation-Dependent Depletion [CADD] [38]). Fig. 2 shows the performance comparison of the pathogenicity prediction tools based on test data from ClinVar release Nov 29, 2020.
All mutations filtered by MedyPatho (MedySapiens) were manually curated by genetic experts.
RESULTS
Clinical patient characteristics
To evaluate the usefulness of the NEOseq_ACTION panel for patients, prospective panel testing was conducted in 111 patients. The clinical characteristics of the patients are shown in Table 2. The mean age at symptom or sign recognition was 10.4 months (range, 1–188 months). Around three quarters of the patients presented with symptoms or showed laboratory abnormalities within a month after birth.
Seventy patients were clinically asymptomatic, and 57 patients were referred to the outpatient clinic because of initial NST abnormality. Patients with NST abnormality were directly enrolled without further investigation, although most of them had simultaneous metabolic screening or NST repetition. Thirteen patients visited the clinic for incidentally detected laboratory abnormalities, such as elevated liver enzymes, creatinine phosphokinase (CPK), or unexplained acidosis. The patients had no perinatal history and showed normal development.
Forty-one patients had one or more clinical symptoms. Neonatal hypotonia and unexplained respiratory failures were the most frequent presentations. However, NST could not provide reliable data because the patients often required mechanical ventilation and total parenteral nutrition because of frequent infections. Among 10 patients with developmental delay, four showed abnormal metabolic screening results, and another four had additional features, including coarse face, skeletal abnormalities, or extensive Mongolian spots suggestive of storage diseases.
Evaluation and validation of the NEOseq_ACTION panel
The average time from patient visit to result reporting was 8.3 days, and the average time from sample preparation to result reporting was 3.4 days. As the MiSeq Reagent Kit v2 Nano was used for less than three samples, sequencing was performed even if only one sample was submitted. We identified significant variants in 10 genes in 15 patients; detailed information is provided in Table 3. Among them, 11 patients were asymptomatic (10 with NST abnormality, and one with abnormal liver function). Four symptomatic patients (neonatal hypotonia, early-onset seizures, skeletal abnormalities, or abdominal distension) were genetically confirmed. Suspected compound heterozygous or homozygous variants were detected in 13 patients (genes related to autosomal recessive diseases), and two hemizygous variants were detected in two patients. Among the 15 patients with suspected recessive diseases, both alleles were initially classified as pathogenic or likely pathogenic in nine patients (P2, P5, P14, P41, P45, P46, P72, P74, and P83). The other patients carried one pathogenic or likely pathogenic allele and one VUS (P6, P10, P25, P36, P40, and P111). Parental testing was performed in four families, and further biochemical testing was performed for all except two patients. Therefore, the final classification was changed for four patients (P6, P25, P36, and P40). The second variant in P10 and P111 remained of unknown significance after retesting. However, the patients started treatment as soon as the biochemical testing results provided strong evidence for the final diagnosis. As P10 and P111 showed a mildly elevated serum phenylalanine level when compared with actual patients with phenylketonuria on repeated testing, their second variants were considered not significant.
Representative cases
All positive results in 15 cases affected the treatment decisions (Table 4). Two patients received a final diagnosis, although they had received appropriate treatment based on clinical suspicion. Eleven patients started medication or changed their daily diet. All families received counseling based on the patients’ results. The average time from testing to medical decision making was 20.8 days (2–68 days). Below, we describe some representative cases.
P2 was a 3.5-year-old girl with no perinatal history. At 12 months, her growth was retarded. At 2 years, she complained of bilateral knee pain, which was followed by gross deformation of several joints. She underwent some tests and received regular rehabilitation, but without clinical improvement. She was referred to our clinic 2 years after the onset of the symptoms and underwent simultaneous NEOseq_ACTION panel sequencing and enzyme testing on suspicion of mucopolysaccharidosis (MPS) type IVA. Compound heterozygous variants in GALNS were identified in 5 days, and glucose 6-sulfatase activity was 0.37 nmol/17 hr/mg protein (reference range [ref], 18.6–61.8). The patient was confirmed to have MPS IVA and started enzyme replacement therapy 3 weeks after the first visit.
P14 was a 3-week-old boy whose NST results indicated low arginine (92.6 μmol/L; ref, 120–600) and high citrulline (172.6 μmol/L; ref, 8–47) levels. The patient was admitted to the neonatal intensive care unit and started a diet with special milk and carglumic acid for 2 weeks. The patient was referred for prompt diagnosis, and panel sequencing demonstrated pathogenic compound heterozygous variants in SLC25A13. He was diagnosed as having citrullinemia type II and started oral arginine therapy. He is currently 13 months old and shows normal development.
P41 was a 4-month-old boy who presented poor suckling and respiratory difficulty after birth. He was lethargic and depended on the nasogastric tube. He could make eye contact with his family, but not control his head. Panel testing indicated pathogenic compound heterozygous variants in PREPL associated with congenital myasthenic syndrome. We attempted repetitive nerve stimulation tests; however, the results were unreliable because of extremely low muscle mass and immature myelination. He started on an acetylcholine esterase inhibitor and salbutamol and showed remarkable clinical improvement. A month after starting the treatment, he could roll and control his head. He started to crawl at 6 months and could stand without support at 13 months. He has speech and eats ordinary meals orally.
P111 was a 1-month-old boy who showed NST abnormality indicating an elevated phenylalanine level (240 μmol/L; ref, 38–137). He underwent NST thrice because of abnormal results at different hospitals. The panel test indicated a pathogenic variant and a VUS in PAH. As detailed metabolic testing indicated slightly increased phenylalanine (100 µmol/L; ref, 25–74) and normal tyrosine (89 μmol/L; ref, 13–91) levels, we concluded that the patient had hyperphenylalaninemia, rather than phenylketonuria. The patient continued on breast milk and showed normal development until the last follow-up.
DISCUSSION
In this proof-of-concept study, we demonstrated the performance of rapid targeted sequencing using a NEOseq_ACTION panel. Actionable genes were identified in 15 patients (13.5%). For actionable genetic diseases, early therapeutic intervention can allow neonates and infants to have a normal life or can minimize the consequences of the disease. We used the NEOseq_ACTION panel to confirm clinically or biochemically suspected diseases. The panel allowed for the diagnosis of various curable or treatable diseases from a few DBSs obtained during routine NST or for critically ill neonates in intensive care unit settings.
Obstacles to the application of rapid WES and WGS in neonates and young infants, including insufficient sequencing depth, limited bioinformatics, and lack of data interpretation experts, have been reported [8]. Multiple factors have to be considered, such as genetic and clinical heterogeneity, including the appearance of formes frustes or incomplete presentation of classical phenotypes in neonates and young infants, and comorbidity because of the fragility of infants. Therefore, a targeted approach covering 100–300 disease-associated genes is cost-effective for rapid detection of medically actionable diseases. A short TAT is essential for the clinical effectiveness of rapid genome sequencing, particularly in neonates.
VUS interpretation can be a bottleneck in reducing the TAT [39]. In this study, rare VUSs with a population frequency of ≤0.6% appeared 25 times per patient (data not shown). As most inborn errors of metabolism are autosomal recessive genetic diseases, clinical geneticists struggle with many VUSs. Therefore, VUSs are the major cause of a prolonged TAT. The initial classification of four variants in four families (P6, P25, P36, and P40) by MedyPatho was changed in the final classification; two VUSs in P6 and P25 were reclassified as likely pathogenic variants based on the results of segregation testing, and two VUSs in P36 and P40 were reclassified as likely pathogenic variants according to biochemical testing results. MedyPatho has shown clinically acceptable accuracy in the classification of clinically relevant variants. Variant classifiers will become more sophisticated as they include accumulating data from specialized panels, such as NEOseq_ACTION, thereby reducing the burden of variant interpretation and the time required for manual curation. Therefore, automatic variant classifiers, such as MedyPatho, are essential to reduce the TAT.
We conducted a prospective study in patients with suspected actionable diseases. More than half of the patients were asymptomatic neonates with NST abnormalities. Although the standard option was to repeat the testing, the patients may have had several unnecessary tests, diet modification, and extra medical expenses. Moreover, we could take immediate, appropriate medical action for confirmed cases. Rapid screening and not missing any treatable diseases are critical to achieve qualitative practice, and the panel provided valuable information to clinicians for patients in the intensive care unit.
This study demonstrated that simultaneous biochemical or functional validation reduced the uncertainty of VUSs. Diseases recently covered in NST panels, such as genetic hearing loss and critical arrhythmias, may require regular check-ups for confirmative diagnosis if patients harbor a VUS. Although numerous ethical and social issues are to be considered, these diseases can be diagnosed and appropriately treated before symptom onset. Accumulating data on VUSs and long-term phenotypes may provide valuable clues on pathogenicity and can be utilized for the advancement of automated analysis programs. For example, P10, referred for NST abnormality, carried missense variants c.975C>G and c.158G>A in PAH. The c.975C>G variant was classified as pathogenic and c.158G>A as a VUS. P10 was diagnosed as having hyperphenylalaninemia with one pathogenic missense variant because serial biochemical testing consistently indicated a mild elevation of the serum phenylalanine level. The c.158G>A variant had no clinical significance. P40 had two variants in MCCC2: c.838G>T was classified as likely pathogenic and c.1342G>A as a VUS. His urine 3-hydroxyisovaleric acid level was 173.1.1 mmol/mol Cr, which is 10 times higher than the normal level, and thus, he was confirmed to have 3-methylcrotonyl-CoA carboxylase 2 deficiency with reclassification of the VUS.
This study demonstrated the importance of collaboration among field experts during panel design. Targeted RGT does not simply require a collection of genes of interest. A multidisciplinary team including clinical experts in pediatrics, clinical geneticists, clinical pathologists, and bioinformaticians should participate in gene selection for confirmative diagnosis. In particular, the details of medical actions should be clearly defined. Such a combined effort can maximize the clinical utility of targeted RGT of medically actionable genes. Moreover, predefining clear medical actions avoids ethical issues.
One of the limitations of this study was the relatively low detection rate. However, considering the benefits of detecting and managing actionable diseases at the presymptomatic stage, our diagnostic yield was acceptable.
NEOseq_ACTION panel sequencing requires only a small amount of DNA, e.g., from DBSs, and the TAT from blood collection to result reporting using the automated analytic pipeline was less than 4 days. The cost of NEOseq_ACTION sequencing is substantially lower than that of rapid WGS or WES, and because automated variant classification systems are embedded in bioinformatics pipelines, fewer experts are required. NEOseq_ACTION can be particularly advantageous in regions where resources and clinical geneticists are scarce, but rare genetic diseases are prevalent.
In conclusion, this prospective pilot study evaluated the clinical feasibility of rapid and timely diagnosis of treatable rare genetic diseases using a NEOseq_ACTION panel. Targeted RGT using the NEOseq_ACTION panel in acutely unwell neonates with suspected genetic diseases represents an example of the clinical application of genomic medicine. Considering the fragility of neonates in medical and ethical terms, the NEOseq_ACTION panel may provide a valuable opportunity for appropriate treatment of presymptomatic or early-stage diseases, even for NST in asymptomatic neonates.
 XML Download
XML Download