

 PDF
PDF Citation
Citation Print
Print



INTRODUCTION
Weaver syndrome (WS; OMIM 277590), first reported in 1974, is a rare congenital disorder characterized by overgrowth, distinct facial features, and accelerated osseous maturation. According to a previous study, WS is caused by a variant in the enhancer of zeste homolog 2 (EZH2) gene [1]. EZH2 variants were also identified in 48 patients with WS with the clinical phenotype described above [1]. Approximately 50% patients were de novo cases, whereas familial cases were inherited in an autosomal dominant pattern [2]. In Korea, although potential WS cases have been reported, an EZH2 variant has not been confirmed [3]. In this case report, we discuss the first Korean case of WS along with genetic confirmation of an EZH2 variant. The Institutional Review Board of Seoul National University Hospital (Seoul, Korea) approved the study (approval number: 2111-131-1275). Informed consent was obtained from the patient’s legal guardians.
Go to : 
CASE
A 7-month-old female was presented to the out-patient pediatrics clinic of Seoul National University Hospital with multiple palpable masses on her back, left side of the head, and extremities. The masses were approximately 1-3 cm in size and moved upon touch, but were reportedly not painful. The patient did not have any perinatal problems and was delivered at full-term. However, her height, head circumference, and weight were ≥97% of those expected for her age. The patient was tall in stature and exhibited macrocephaly. Physical examinations revealed doughy skin and umbilical hernia; however, camptodactyly, hoarse cry, and abnormal tone were absent. Her characteristic facial features included frontal bossing, round face, retrognathia, wide-spaced eyes, low-set ears, and cleft chin (Table 1). In the magnetic resonance imaging examination, a 7 cm mass was found on the left adrenal gland. Pathological examination of the mass revealed neuroblastoma on the left adrenal gland, and bone marrow aspiration confirmed the presence of neuroblastoma cells in the bone marrow (Fig. 1A).
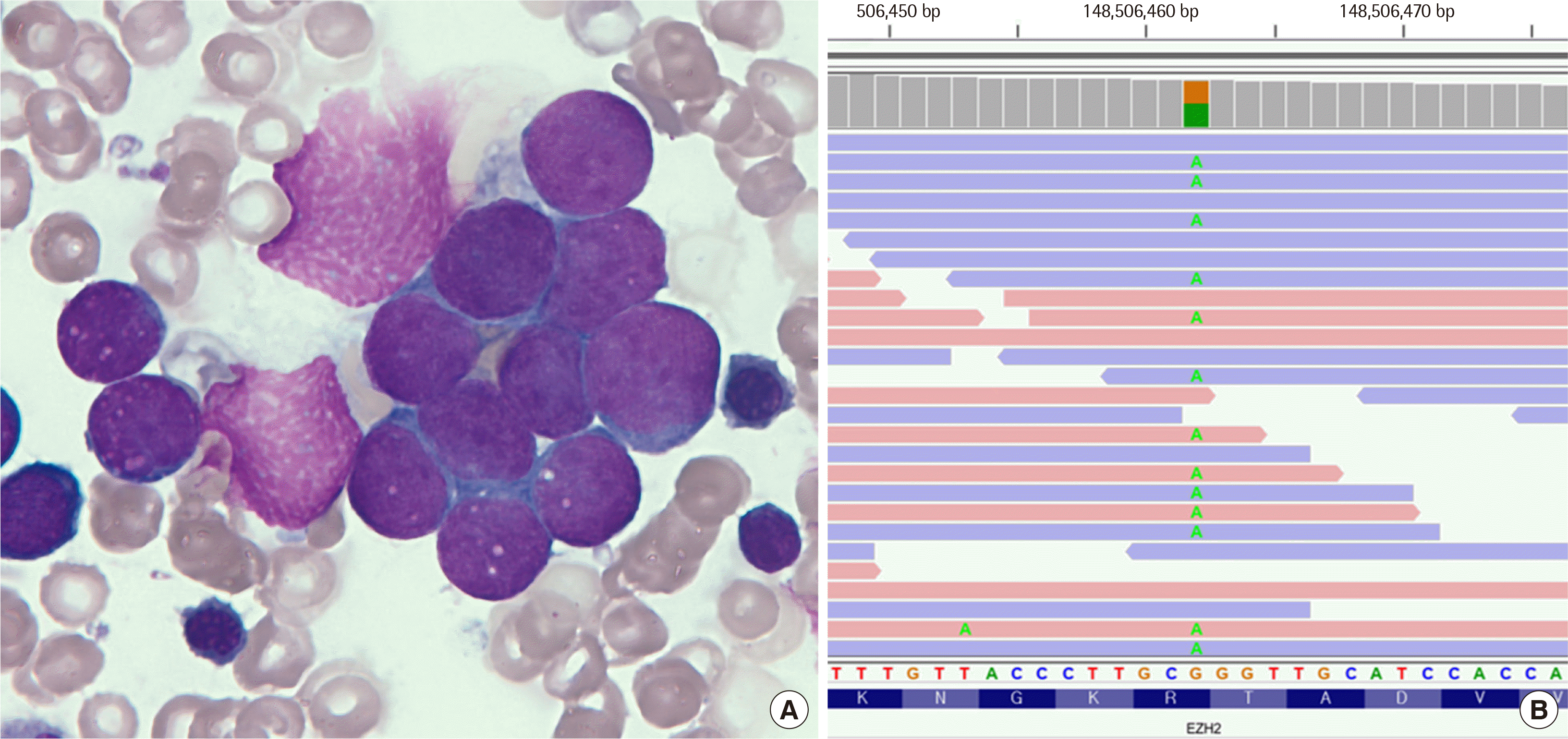 | Fig. 1Morphological characteristics of bone marrow aspirate and visualization of next-generation gene sequencing data of the patient. (A) Neuroblastoma cells cluster with fibrils (Wright-Giemsa stain, ×1,000). (B) Sequence visualization of enhancer of zeste homolog 2 (EZH2) gene showing a missense variant, c.2050C>T.
|
Table 1
Comparison of clinical phenotype of our patient with those of previous cases of Weaver syndrome
| Current study | Case 1 [14] | Case 2 [22] | Case 3 [23] | n/N* (%) [24] | n/N* (%) [2] | |
|---|---|---|---|---|---|---|
| Facial features† | + | + | + | + | 3/3 (100) | 31/48 (65) |
| Hypertonia | - | - | + | - | 1/3 (33) | 11/39 (28) |
| Hypotonia | - | - | - | - | 2/3 (67) | 18/41 (44) |
| Camptodactyly | - | - | - | + | 2/3 (67) | 17/38 (45) |
| Umbilical hernia | + | - | + | - | 3/3 (100) | 17/40 (43) |
| Doughy skin | + | - | - | - | 2/3 (67) | 17/35 (49) |
| Hoarse cry | - | - | - | - | 3/3 (100) | 10/27 (37) |
| Malignancy | NB | AML | - | - | 0/3 (0) | 2/48 (4)‡ |
| EZH2 | ||||||
| AA change | R684C | P132L | E745K | H240R |
*n/N refers to the proportion of patients exhibiting the corresponding phenotype among the total number of patients included in the reference reports describing the presence of a specific phenotype. †Facial features characterizing WS, namely ocular hypertelorism, frontal bossing, round face, and retrognathia, ‡Three types of cancer occurred in two patients with WS (lymphoma, NB and ALL).
![]()
As the patient was suspected of having overgrowth syndrome, she underwent next-generation sequencing-based gene panel testing, which revealed a heterozygous missense variant in NM_004456.4 (EZH2):c.2050C>T (p.Arg684Cys) (Fig. 1B). This variant is absent from large population databases such as gnomAD (https://gnomad.broadinstitute.org/) and the Korean Reference Genome Database (http://coda.nih.go.kr/coda/KRGDB/). A previous functional study demonstrated that, in the presence of the c.2050C>T (p.Arg684Cys) variant of EZH2, the histone methyltransferase activity of the polycomb repressive complex-2 (PRC2) protein was significantly decreased compared to that of the wild-type enzyme [4]. In addition, a different missense change in the same codon (p.Arg684His) has been reported as a pathogenic variant [2]. In silico analyses using SIFT, Polyphen-2, and Mutation Taster supported the finding that this variant has deleterious effects on the protein function. This variant was previously reported as pathogenic [2, 4, 5]; thus, we subsequently classified it as pathogenic (PS3, PM2, PM5, PP3, and PP5) based on the 2015 American College of Medical Genetics and Genomics and Association for Molecular Pathology guidelines [6]. Other genes in the panel, including AKT3, BRWD3, DNMT3, EED, GPC3, HIST1H1E, MTOR, NFIX, NSD1, PIK3CA, PPP2R5D, and PTEN, did not contain variants. Moreover, the EZH2 variant was detected in the adrenal gland biopsy tissue, and as a result, the patient was diagnosed with WS. She was started on chemotherapy with etoposide and doxorubicin, and her neuroblastoma showed a partial response after 10 cycles of treatment.
Go to :
DISCUSSION
EZH2 encodes a histone-lysine N-methyltransferase that is a component of PRC2, which is associated with epigenetic silencing in cells. PRC2 exhibits histone methyltransferase activity and methylates histone H3 on lysine 27; the EZH2 variant causes a partial loss of this methyltransferase function [7]. Variants in other PRC2 elements or other histone methyltransferase-related genes are genetically distinct from WS; however, some cases exhibit an overgrowth phenotype [8]. Particularly, these mechanisms are similar to the impaired histone methyltransferase activity of the variant of nuclear receptor binding SET domain protein 1 (NSD1), which causes Sotos syndrome, a type of overgrowth syndrome [9]. Sotos syndrome shows a high level of phenotypic overlap with WS [10]; therefore, overgrowth is expected to occur in cases of impaired methylation regulation [11]. However, analysis of EZH2 protein in patients with WS did not reveal a clear correlation between the severity of clinical symptoms and greatly decreased histone methyltransferase activity [4]. Therefore, phenotypic differences among patients with WS may be related to additional factors.
Both somatic gain-of-function and loss-of-function mutations of EZH2 are associated with solid tumors, such as breast and prostate cancer, or hematologic cancers [12]. In contrast, evidence for the increased risk of malignancy in patients with EZH2 germline variants is not sufficient [13]. Previous studies reported that cancers such as neuroblastoma, acute myeloid leukemia, acute lymphoblastic leukemia, teratoma, ovarian tumor, and lymphoma occur at an early age in patients with WS [1, 2, 14]. Three cases of neuroblastoma have been reported in 3-, 8-, and 9-month-old patients with WS, whereas only one case has been reported for each of the other cancer types [2]. Our patient was 6 months old at the time of neuroblastoma onset (7 months old when she presented to the clinic), which is consistent with similar cases. This case supports the finding that germline variants in EZH2 may be associated with cancer, specifically neuroblastoma [15].
Diagnosing overgrowth syndrome is important for providing genetic counseling for cancer surveillance and prognosis [16]. Particularly, patients with Beckwith-Wiedemann syndrome show an increased risk of Wilms’ tumor [17]. The incidence of cancer is reportedly increased in patients with other overgrowth syndromes, and variants in genes such as DNA (cytosine-5)-methyltransferase 3A (DMNT3A) are associated with solid or hematologic cancer [18-20]. The regular cancer screening plan has been proposed for patients with theses syndromes [21]. As neuroblastoma occurs increasingly in WS, we recommend that patients with WS should also undergo cancer surveillance.
This is the first Korean case of genetically confirmed WS, accompanied by the rare occurrence of early onset neuroblastoma. Although EZH2 variant-induced overgrowth syndrome is rare, it should be included in the gene panel when evaluating overgrowth syndrome. Moreover, if overgrowth is accompanied by neuroblastoma, the probability of WS is high, and genetic testing for germline variants can help to confirm this diagnosis.
Go to :
 XML Download
XML Download