

 PDF
PDF Citation
Citation Print
Print



INTRODUCTION
Mesenchymal hamartoma of the liver (MHL) is the second most common benign pediatric tumor, although its incidence is as low as 8% of all pediatric malignancies [1]. Moreover, 80% of patients are diagnosed before the age of 2 years, while the remaining 20% are diagnosed before the age of 5 years [2,3]. The occurrence of MHL in the adult population is very rare. A literature review by Klaassen et al. [4] presented only 31 published adult cases until 2010 [5]. MHL usually manifests as a cystic mass in the liver that occupies the intraperitoneal cavity and presents with vague symptoms. It is presumed that abnormal development of a complex mixture of immature mesenchymal tissues in the portal tract contributes to its pathogenesis. MHL is considered a benign tumor, and few studies have reported spontaneous resolution of the tumor [6,7]. The correct management of MHL remains controversial, but complete excision is recommended [8]. Complete resection usually achieves good outcomes without tumor recurrence, [8] which was also demonstrated in a retrospective study of nine patients followed up for 6.00 ± 2.78 years (range: 1–9 years) [9]. However, the prognosis of untreated tumors is not well understood. Several studies have reported about the development of undifferentiated embryonal sarcoma (UES) or angiosarcoma secondary to incompletely resected MHL [6,10]. Since the primary pathogenesis is dysgenesis of the portal tract, including the bile duct, and progression from bile duct hamartoma to intrahepatic cholangiocarcinoma (iCCA) has been reported in several studies, iCCA should be included in the differential diagnosis of malignant transformation of MHL [11,12]. These facts underline the importance of complete surgical resection and histopathological diagnosis. Here, we have presented a novel case of iCCA that developed 24 years after the initial diagnosis of MHL.
Go to : 
CASE
A 26-year-old female was admitted for MHL. At the age of 16 months, a mass measuring 9 cm × 8 cm with multiple septations in the right lobe of the liver was noted during the assessment of abdominal distension using ultrasonography (US) and magnetic resonance imaging (MRI) (Fig. 1A, 1B). Based on these findings, the patient was diagnosed with MHL. Liver enzyme levels were elevated, but not above three-fold of the upper limit. The serum α-fetoprotein (AFP) level was elevated to 39 mg/mL, while the carcinoembryonic antigen (CEA) level was 3.3 ng/mL. However, the scheduled hepatic mass resection was cancelled due to an upper respiratory infection. Since this mass did not cause any symptoms, the patient underwent regular sonographic follow-up every 2 years until the age of 10 years. Subsequently, the patient was lost to follow-up. At the age of 26 years, the patient returned to our clinic with abdominal pain lasting for 6 weeks. Computed tomography (CT) revealed an increase in mass size to 12.9 cm × 8.3 cm and a newly developed peripheral enhancing lesion in its posterior portion measuring 6 cm (Fig. 1C). Follow-up MRI and 18F-fluorodeoxyglucose positron emission tomography (PET) could not exclude malignant transformation (Fig. 1D, 1E). In addition, enlarged lymph nodes (LN) in the hepatoduodenal ligament and portocaval space were also identified. Her liver function appeared to be normal. Mild elevation of the γ-glutamyl transpeptidase level to 40 IU/L (normal range: 8–35 IU/L) was observed. Preoperatively, the serum levels of tumor markers were as follows: AFP: 1.13 ng/mL (normal range: 0.89–8.78 ng/mL), CEA: 1.3 ng/mL (normal range: 0–5 ng/mL), and carbohydrate antigen 19-9 (CA 19-9): 1,075 U/mL (normal range: 0–37 U/mL). Mass excision with segment 5 and 6 hepatic segmentectomy (non-anatomical) and en bloc hilar LN dissection was performed. The mass was approached via an upper midline incision, and a mass measuring 16 cm was excised with a part of segments 5 and 6 of the liver parenchyma. Intraoperative frozen biopsy of the hilar LN showed no evidence of a tumor. Histopathological examination of the mass measuring 12.0 cm × 11.0 cm × 8.0 cm confirmed the diagnosis of iCCA in the background of MHL, with a T2N0M0 pathological stage (stage II). Although lymphatic and venous invasions were noted, the resection margin was free from tumor cells. Immunohistochemical analysis revealed cytokeratin (CK) 19 and p40 positivity and loss of p53 expression; thus, the final diagnosis was iCCA with squamous differentiation (Fig. 2). The patient recovered well postoperatively; however, a biloma was observed at the resection site, which required percutaneous drainage for 29 days. Furthermore, adjuvant chemotherapy (5-fluorouracil, 810 mg, six cycles) was initiated on postoperative day 60. After surgery and adjuvant chemotherapy, the serum levels of CEA and CA 19-9 decreased to 1.6 U/mL and 7 ng/mL, respectively (Fig. 3).
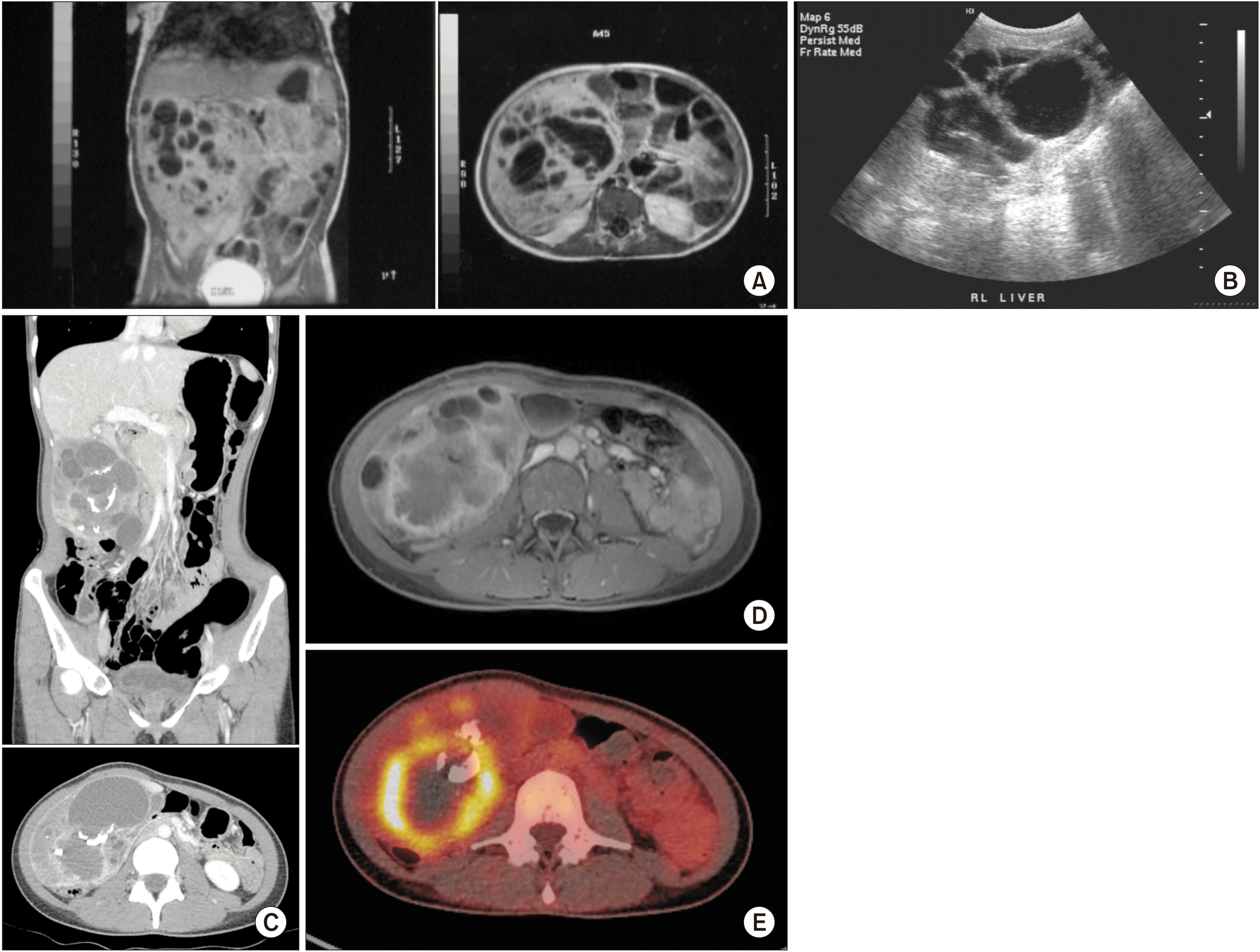 | Fig. 1Right hepatic mass in imaging findings (A) Magnetic resonance imaging (MRI) at the initial diagnosis (at 16 months of age). On coronal view, a cystic mass measuring 9 cm × 8 cm is observed in the right lobe of the liver. (B) Follow-up ultrasonography (at 5 years old). Although the limitation of the test precludes an accurate evaluation of size, continuous monitoring shows that the mass with multiple cysts does not exhibit a rapid increase in size. (C) Computed tomography before the operation. A complex solid, cystic mass with thick wall and calcification in the liver segment 6 is observed along with an increase in size (12.9 cm × 8.3 cm). In the portal phase, a peripheral enhancing lesion measuring 6 cm is seen in the posterior portion. This indicates the possibility of abscess formation or malignant transformation to undifferentiated embryonal sarcoma. (D) MRI before the operation. In the T2 enhanced image, the lesion is observed with a solid component and peripheral rim-like enhancement along the posteroinferior portion of the mass. (E) F-18-fluorodeoxyglucose positron emission tomography (PET). The rim-like enhancement on MRI shows intense hypermetabolism on PET.
|
 | Fig. 2Diagnosis of intrahepatic cholangiocarcinoma (iCCA) in the background of mesenchymal hamartoma of the liver (MHL). On gross pathology, a mass (size: 12.0 cm × 11.0 cm × 8.0 cm) with a necrotic portion (10%) and calcification is observed. Histopathology reveals iCCA in the background of mesenchymal hamartoma and indicates the presence of adenosquamous carcinoma with intrahepatic vascular invasion. Lymphatic and venous invasions are confirmed microscopically, although no perineural invasion is observed. Immunohistochemistry shows expression of cytokeratin (CK) 19 and p40 and loss of p53. This finding suggests a cancer with squamous differentiation. (A) Left ×2: Gross photos. Middle: H&E stain ×40. Right: H&E stain ×40. (B) CK19 statin (×40). (C) p40 stain (×40). (D) p53 stain (×40).
|
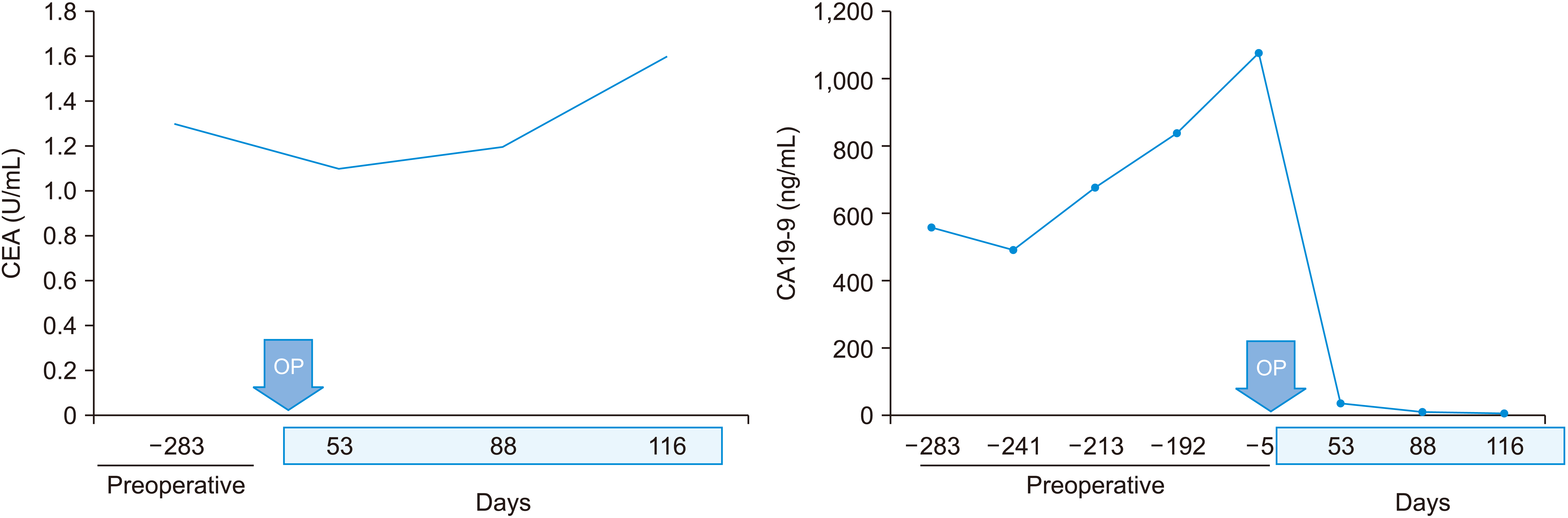
This study was approved by the Institutional Review Board of Seoul National University Hospital (2202-107-1302). Informed consent was obtained from the patient for data collection and publication of the study.
Go to :
DISCUSSION
MHL is considered a benign tumor that mainly presents in the right lobe of the liver. It typically occurs in children aged less than 2 years [13]. Although rare, MHL has been reported in older children and adults [4,5]. However, the reported adult cases provided insufficient information regarding whether the mass developed at the time of diagnosis or during childhood.
The clinical manifestations of MHL are diverse, ranging from painless abdominal distension to compromised liver function and symptoms (e.g., abdominal pain) due to an enlarged mass. In our case, an enlarged mass was incidentally found during work-up at 16 months of age. The patient had been asymptomatic for 24 years, developing abdominal pain only shortly before surgery.
Surgical resection is the treatment of choice in most review articles. Nevertheless, when a clinician or a patient decides not to operate MHL, the authors recommend a check-up with images and tumor markers (CA19-9 and CEA) at an interval of 6–12 months. However, there is no concrete evidence for the authors’ opinion.
US and CT are the diagnostic modalities of choice because they allow proper inspection of gross tumor features. On diagnostic imaging, MHL presents as a solitary mass, predominantly cystic, but it can present as a cystic, multicystic, or complex solid mass [14]. Cystic masses are visualized as multicystic and septated masses within the liver or on a pedicle. Low and high signal intensities are characteristic findings on T1- and T2-weighted MRI. Calcification, occasionally multifocal, may also be present. In contrast studies, the characteristic findings include enhancement of the solid component, abnormal blood vessels, and multiple focal avascular lesions in the cystic portion. The multiseptated mass in our patient persisted since the initial diagnosis, suggesting a cystic form of MHL, similar to the findings of follow-up US at the age of 5 years. However, CT performed at the age of 26 years revealed a rim enhancement inside the mass, arousing suspicion of abscess formation and malignant transformation, which was confirmed by subsequent MRI and PET. Since the patient was an adult with persisting MHL from 2 years of age, it was difficult to rule out malignant transformation; therefore, surgical treatment was considered as the first treatment option. Several studies have reported that UES can arise from incompletely resected MHL [8,10]. Recent studies have shown that MHL is associated with changes on chromosome 19, and it shares this genetic profile with UES secondary to MHL (rearrangement in the 19q13.4 region) [15]. However, none of the studies have reported an association between MHL and other cancers thus far.
The pedunculated mass was excised with a part of the liver parenchyma since complete excision was considered the best treatment option. Aspiration cytology performed during preoperative diagnosis mainly identified benign spindle cells mixed with myxoid stroma, normal ductal cells, and hepatocytes. Mesenchymal structures containing abnormal bile duct profiles and bile duct equivalents surrounding immature, partly myxoid mesenchyme with spindle cells and stellate cells are often seen in MHL [13]. Since microscopic examination of our patient’s material showed similar features, we concluded that the mass was MHL. Although these examinations were not performed due to the possibility of malignant transformation, we found evidence suggesting iCCA. Positive staining for vimentin and smooth muscle actin suggests MHL, whereas positive staining for CK-7 and CK-19 indicates the presence of bile duct-like structures. The expression of CK-19 in our patient indicated abnormal proliferation of bile ducts. iCCA is an adenocarcinoma arising from the intrahepatic bile ducts [16]. Loss of p53 expression implied malignant transformation. In addition, p53 is a diagnostic marker for iCCA [17]. Therefore, we concluded that iCCA developed in the background of MHL. Postoperative treatment was performed according to the guidelines for iCCA stating that complete resection is sufficient for MHL.
Abnormal development of the portal tract, including the bile duct, is the primary cause of MHL. Moreover, progression from bile duct hamartoma to iCCA has been reported in previous studies [11,12]. Although the mechanism and histopathological association between MHL and bile duct hamartoma have not been uncovered since both hamartomas include bile duct structures, iCCA should be considered in the differential diagnosis when malignant transformation of MHL is suspected. Thus, en bloc surgical resection with negative surgical margins and histopathological diagnosis are more important.
MHL is a rare disease, with limited information available on its pathogenesis or long-term course. Here, we have reported a novel case of iCCA secondary to untreated MHL in a 26-year-old patient. Our findings differ from those of previously published reports on malignant transformation of MHL, such as progression to UES or angiosarcoma. The finding suggests that more aggressive surgical treatment is required for MHL. Early resection of MHL is recommended because of malignant transformation even in a child under 2 years of age. We anticipate that our finding will impact the treatment guidelines for MHL in the future.
Go to :
 XML Download
XML Download