

 PDF
PDF Citation
Citation Print
Print


Introduction
The skin often suffers from acute or chronic injuries, such as diabetic ulcers and accidental skin injuries (blunt force tears or extensive burns) (1). The repair of a skin wound is orchestrated by the cooperation of cell migration, proliferation, differentiation, collagen deposition, angiogenesis, and remodeling (2). In some pathological diseases, the speed of wound healing will be reduced because of interference, which leads to chronic non-healing wounds or the formation of pathological scars (3). To speed up wound healing, several treatment modalities, including cytokines/growth factors and cell-based therapies, are currently used, with stem cell therapy being suggested as a potential treatment option for non-healing wounds (4, 5).
Mesenchymal stem cells (MSCs) are a population of self-renewing cells that are able to accelerate wound healing (4). These cells can be recruited into the bloodstream after injury and engrafted into the remodeling microvasculature (6). However, the direct use of stem/progenitor cells is restricted by the potential clinical side effects (7). Their paracrine functions, including the secretion of growth factors, cytokines, and exosomes, have been noticed for decreasing the risk of MSC-based therapies (8). A previous study reports that MSCs improve tissue repair depending on exosome secretion (9). It has been shown that exosomes derived from MSCs (MSCs-Exos) have the advantage of avoiding mutation or transfer of cells that damage DNA, easily circulating through capillaries, and maintaining high relative concentration in a relatively short time (8). In the animal model of diabetes or skin burn, exosome injection can activate the angiogenesis and promote the propagation and migration of skin cells and the wound closure process (7, 10). This suggests that exosomes are valuable therapeutic tools for wound healing. However, scarce in-depth study on its specific molecular mechanism limits its clinical implementation.
Exosomes are vesicles able to transport proteins, microRNAs (miRNAs), and mRNAs between cells (11). A number of studies have revealed that exosomes can exert their pro-healing effect on animals with skin wounds through shuttling cargoes (12-14). MiR-150-5p could be altered by small extracellular vesicles at the molecular level in the wound healing process (15), while the mechanisms by which miR-150-5p mediates skin wound repair have not been reported yet. In 2011, Lin Cao and colleagues (16) found that decreased phosphatase with tensin homology (PTEN) expression at the edges of damaged corneal epithelium increased the phosphatidylinositol 3-kinase (PI3K)/AKT pathway, which promotes cell migration and wound healing. Leszczynska et al. (17) found that exosomes were a promising candidate for the treatment of gelled cell injury in diabetic patients, possibly by activating the AKT pathway. Herein, we boldly hypothesized that exosomal miR-150-5p could affect skin wound healing via PTEN-mediated PI3K/AKT pathway since miR-150-5p was reported to downregulate PTEN and promote the growth and migration of colon cancer cells (18). The aim of this work was to explore the molecular mechanism of the MSCs-Exos in promoting skin wound healing, so as to provide innovative and optimal solutions for clinical problems of wound healing in the future.
Go to : 
Materials and Methods
Isolation and culture of MSCs
Human umbilical cord MSCs (HUC-MSCs) (Shanghai Honsun Biotechnology Co., Ltd.) were immersed in 10% fetal bovine serum (FBS)-supplemented Dulbecco’s modified Eagle medium (DMEM) in a 5% CO2 incubator at 37℃ and the culture medium was changed every three to four days. When the HUC-MSCs covered the bottom of the culture bottle, that is, when the cell confluence reached 80%∼90%, the cell passage was carried on at a ratio of 1:3.
Identification of HUC-MSCs
The adherent HUC-MSCs were passaged and assayed at passage 3 (P3). The P3 cells were added with osteogenic medium (Cyagen Biosciences Inc., Guangzhou, China) and chondrogenic medium (Cyagen Biosciences Inc.) for 21-day cell culture and with adipogenic medium (Cyagen Biosciences Inc.) for 14-day cell culture. After the cell culture, the osteogenic, chondrogenic, and adipogenic differentiation abilities of the HUC-MSCs were respectively identified by alizarin red staining (Shanghai Yuanye Biotechnology Co, Ltd., Shanghai, China), Alcian blue staining (Shanghai Yuanye Biotechnology Co, Ltd.), and oil red O staining (Shanghai Yuanye Biotechnology Co, Ltd.), and the cells were then observed under a microscope and photographed. Flow cytometry was used to examine the expression of CD34 (a negative marker for stem cells), CD29, CD90, and CD44 (positive markers for stem cells) in the P3 adherent cells.
MSCs modified by miR-150-5p
MiR-150-5p overexpression (miR-150-5p-mimic) and inhibition (miR-150-5p-inhibitor) lentiviral vectors and their negative controls (NCs, mimic-NC, inhibitor-NC) were provided by Hanbio Biotechnology Co., Ltd. (Shanghai, China). The HUC-MSCs were disassociated, passaged, and cultured overnight before lentiviral infection (plural 100). Forty-eight hours after the infection, the supernatants were collected to prepare HUC-MSCs-derived exosomes (MSCs-Exo). Exosomes derived from HUC-MSCs infected with miR-150-5p mimic, miR-150-5p inhibitor, or their NCs were termed as MSCs-Exo-mimic-NC, MSCs-Exo-miR-150-5p-mimic, MSCs-Exo-inhibitor-NC, and MSCs-Exo-miR-150-5p-inhibitor. Green fluorescent protein was used as a marker for measuring the infection efficacy. The efficacy of miR-150-5p overexpression and inhibition lentiviral vectors was verified by reverse transcription-quan-titative polymerase chain reaction (RT-qPCR).
Collection and isolation of MSCs-Exos
When MSCs adhered to the wall and grew to about 80%, they were passaged at the ratio of 1:3. The medium was renewed for the first time after 24 hours and then was changed every 3 days. MSCs at P3-P4 were routinely cultured into 80%∼90% confluence and then the medium was replaced with FBS-free DMEM (Thermo Fisher Scientific, Waltham, MA, USA) for 2-day culture, followed by the collection of the supernatant. The cells adhered to the wall and grew all over the bottom of the cell flask. When the cell confluence reached 80%, the HUC-MSCs were passaged at a ratio of 1:3, and the medium was renewed, during which the supernatant was collected. The collected supernatant was stored in a refrigerator at −20℃. When enough supernatant was collected, the supernatant was thawed and filtered with a 0.22 μm filter to remove impurities. The collected supernatant was centrifuged for 70 minutes (100,000 g, 4℃), after which the supernatant was discarded and the bottom and sidewalls of the centrifuge tube were washed with 1 ml phosphate-buffered saline (PBS). The pellet was resuspended. The protein concentration was quantified by the bicinchoninic acid (BCA) method and recorded in detail. The suspended pellet was stored in a −80℃ refrigerator for follow-up experiments. The morphology and size of the pellet were observed by a transmission electron microscope (TEM). CD63 and CD81 protein levels were detected using western blot analysis.
Keratinocyte culture and transfection
HaCaT cells from China Center for Type Culture Collection, Wuhan University were cultured in exosome-free DMEM (HyClone, Logan, UT, USA) containing 10% FBS (Gibco, Carlsbad, CA, USA) in a wet incubator (37℃, 5% CO2).
Cell transfection was carried out according to the specification of the Lipofectamine 3000 reagent (Invitrogen, Carlsbad, CA, USA). Short hairpin RNA targeting PTEN (sh-PTEN, 50 nM), pCDNA3.1-PTEN (2 μg), and the NCs (sh-NC, pcDNA3.1) were purchased from GenePharma (Shanghai, China).
HaCaT cells were divided into 10 groups, including PBS group (without transfection or exosome incubation), MSCs-Exo group (48-h incubation with 50 μg/ml MSCs-Exos), H2O2 group (24-h induction with 300 μM H2O2), H2O2MSCs-Exo group (48-h incubation with 50 μg/ml MSCs-Exos prior to 24-h induction with 300 μM H2O2), H2O2MSCs-Exo-NC group (48-h incubation with 50 μg/ml MSCs-Exo-NC prior to 24-h induction with 300 μM H2O2), H2O2MSCs-Exo-miR-150-5p group (48-h incubation with 50 μg/ml MSCs-Exo-miR-150-5p before 24-h induction with 300 μM H2O2), H2O2MSCs-Exo-NC+NC group (trans-fection with pcDNA3.1 before 48-h incubation with 50 μg/ml MSCs-Exo-NC and 24-h induction with 300 μM H2O2), H2O2MSCs-Exo-miR-150-5p+NC group (transfection with pcDNA3.1 before 48-h incubation with 50 μg/ml MSCs-Exo-miR-150-5p and 24-h induction with 300 μM H2O2), H2O2MSCs-Exo-NC+PTEN group (transfection with pc DNA 3.1-PTEN before 48-h incubation with 50 μg/ml MSCs-Exo-NC and 24-h induction with 300 μM H2O2), and H2O2MSCs-Exo-miR-150-5p+PTEN group (transfection with pcDNA3.1-PTEN before 48-h incubation with 50 μg/ml MSCs-Exo-miR-150-5p and 24-h induction with 300 μM H2O2).
Cell counting kit-8 (CCK-8) assay
The inhibitory effect of H2O2 on keratinocytes was investigated by using CCK-8 (Beyotime, Shanghai, China). HaCaT cells were seeded onto a 96-well plate containing a serum-free medium (5,000 cells/well) and then exposed to different concentrations of H2O2 (0∼500 μM) for 4 hours.
On the other hand, CCK-8 was used to detect the effect of exosomes on the viability of HaCaT cells. HaCaT cells were seeded onto 96-well plates containing a serum-free medium (2,000 cells/well), and then MSCs-Exo, H2O2MSCs-Exo, H2O2, or the equal amount of exosome diluent (PBS) were added and cultured with HaCaT cells for 1, 2, or 3 days. Then, CCK-8 (10 μl/well) solution was added for the detection of cell viability. The optical density (OD) at 450 nm was measured using Multiskan FC (Thermo Fisher Scientific).
Flow cytometry assay
An Annexin V-fluorescein isothiocyanate cell apoptosis kit (Beyotime) was used to detect keratinocyte apoptosis. To investigate the effect of H2O2 on the apoptosis of HaCaT cells, H2O2 (0∼500 μM) was added and incubated with HaCaT cells for 4 hours.
To investigate the effect of MSCs-Exo on the apoptosis of HaCaT cells, HaCaT cells were cultured with MSCs-Exo, H2O2MSCs-Exo, H2O2, or PBS for 24 hours. The apoptosis rate of HaCaT cells was measured by flow cytometry assay (BD Biotechnology, San Jose, CA).
Detection of cell migration by scratch test
HaCaT cells (7.5×104 cells/well) were seeded onto 24-well plates in DMEM containing 10% serum for 48-hour cell culture and then incubated in serum-free DMEM for 24 hours. Next, the serum-free DMEM was removed and a sterile pipette was used for scratching. After PBS washing, MSCs-Exo, H2O2MSCs-Exo, H2O2, or PBS were added into DMEM containing 1% serum and 1 µM sub-lethal concentration of anti-proliferative agent mitomycin C (MMC, S8146, Selleckchem) for 0-hour and 24-hour incubation (37℃, 5% CO2) and photographed respectively. The scratch area was analyzed using the Image J software, and the wound healing rate was calculated.
RNA immunoprecipitation (RIP)
RIP experiments were run as described in previous literature (19). Briefly, 2×107 HaCaT cells were used to perform RIP experiments using an anti-Argonaute 2 (Ago2) antibody (ab186733, Abcam, Cambridge, UK). Cells of each group were treated with 5 μg Ago2 antibodies for each RIP, with normal rabbit IgG (Abcam, ab125900) as the NC. Then co-precipitated RNAs were extracted and detected by RT-qPCR analysis.
Wound healing rate
The wound healing rate was measured by Image-pro Plus 6.0 software and calculated based on the following formula: Wound healing rate=(Original wound area-Un-healed wound area)/Original wound area×100%.
RT-qPCR
Total RNA was isolated with TRIzol reagent (Invitro-gen), and then reversely transcribed into cDNA by a reverse transcription kit (TIANGEN, Beijing, China). The primers used in real-time fluorescent qPCR were synthesized by Sangon (Shanghai, China) according to the sequences shown in Table 1.
A total of 10 μl cDNA was amplified by real-time fluorescent qPCR with SYBRⓇ Green Real Time PCR Master Mix (Roche Diagnostics, Indianapolis, IN) on a Roche 480 real-time PCR system. MiR-150-5p relative expression and PTEN relative mRNA expression were measured by the ΔΔCt method, with U6 or glyceraldehyde-3-phosphate dehydrogenase (GAPDH) as the internal reference.
Western blot analysis
The cells were weighed and homogenized with the Radio-Immunoprecipitation assay lysis buffer (Beyotime) containing protease inhibitor for protein extraction. The protein content was measured by the BCA method (Beyo-time), and an appropriate volume of 5×sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) loading buffer was added.
A total of 15 μl protein sample was added to each well, and 10% SDS-PAGE was performed at 300 mA. Then the protein was transferred onto a polyvinylidene fluoride membrane, and 5% skimmed milk powder was used to seal the membrane at room temperature for 1 hour.
The membrane was probed with primary antibodies against CD63 (ab134045, 1:4,000; Abcam), CD81 (ab79559, 1:1,000; Abcam), PTEN (ab31392, 1:1,000; Abcam), PI3K (ab140307, 1:1,000; Abcam), AKT [#4685S, 1:1,000; Cell Signaling Technology (CST), Danvers, MA, USA], p-PI3K (ab182651, 1:1,000; Abcam), p-AKT (#4060S, 1:1,000; CST), calnexin (CANX, ab22595, 1:1,000; Abcam), Golgi matrix protein 130 (GM130, ab52649, 1:5,000; Abcam), and Histone H3 (ab1791, 1:4,000; Abcam) overnight on a shaking table at 4℃. After washing with Tris-buffered saline with Tween 20 (TBST), secondary antibodies were incubated with the membrane at room temperature for 1 hour. After TBST washing, the chemiluminescence liquid was dripped evenly. Following color development, Image J was used to analyze the result, with GAPDH as the internal reference.
Dual-luciferase gene assay
The binding sites of miR-150-5p to PTEN were predicted by the starBase tool. Based on the predicted results, wild-type (WT) and mutant (MUT) sequences (WT-PTEN and MUT-PTEN) of the PTEN 3’-untranslated region (3’UTR) binding sites were designed and synthesized, respectively. The WT and MUT sequences of PTEN-3’UTR were cloned into single pmirGLO vectors to construct the reporter plasmids of pmirGLO-PTEN-WT and pmirGLO-PTEN-MUT. The reporter plasmids pmirGLO-PTEN-WT and pmirGLO-PTEN-MUT [pmirGLO vectors containing WT-PTEN or MUT-PTEN) were both constructed by Promega (Madison, WI, USA). The two reporter plasmids were co-transfected with miR-150-5p mimic into primary 293T cells (Shanghai Beinuo Biotechnology Co., Ltd., Shanghai, China). After transfection for 48 hours, the activities of firefly and Renilla luciferase were measured on a fluorescence luminescence detector (Promega) according to the instructions of the Dual-Luciferase Reporter Gene Assay Kit (Beyotime). The ratio of target luciferase activity/internal reference luciferase activity was used as the relative luciferase activity to calculate the luciferase activity. The experiment was repeated three times.
Statistical analysis
Data were analyzed with SPSS 18.0 (IBM Corp., Armonk, NY, USA) and GraphPad Prism 7.0 (GraphPad Software Inc., San Diego, CA, USA). The measurement data were presented in the form of mean±standard deviation. Comparisons between the two groups were performed by the T-Test, and comparisons among multiple groups were analyzed by one-way analysis of variance. p< 0.05 was regarded as a statistically significant difference.
Go to :
Results
Isolation and identification of the MSCs-Exos
Under the inverted microscope, the primary HUC-MSCs were distributed loosely and were in round, with good refraction. The adherent HUC-MSCs were passaged when the confluence reached 80%∼90%. The passaged cells grew stably, and most of the cells were present in a spindle shape (Fig. 1A). The P3 HUC-MSCs were induced for multi-directional differentiation. Following the 14-day adipogenic induction, the HUC-MSCs were stained with oil red O. A large number of orange-red lipid droplets were observed in the cells (Fig. 1B). Alizarin red staining was performed after the 21-day osteogenic induction, which showed calcified nodules in the HUC-MSCs (Fig. 1C). After 21 days of culture for chondrogenic induction, Alcian blue staining results showed the formation of proteoglycan, which indicated the potential for chondrogenic differentiation (Fig. 1D). Cell surface antigen markers were detected by flow cytometry. The results showed that the isolated HUC-MSC were positive for CD29, CD90, and CD44, but did not express CD34 (Fig. 1E). All these results showed that the isolated cells shared the characteristics of MSCs.
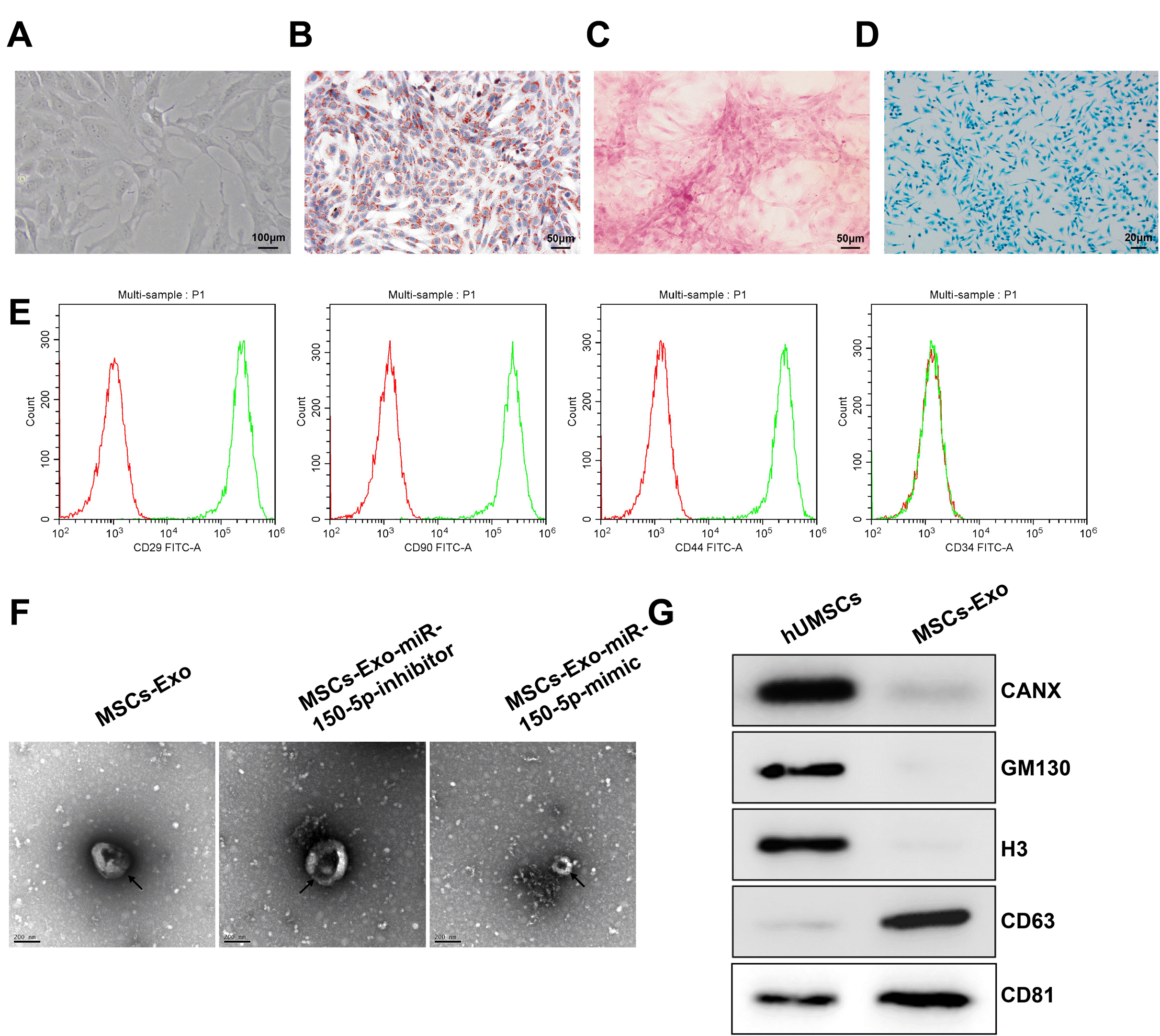 | Fig. 1Isolation and identification of the MSCs-Exos. (A) The morphology of primary MSCs under an inverted microscope. (B) Oil Red O staining after adipogenic differentiation. (C) Alizarin red staining after osteogenic differentiation. (D) Alcian blue staining detecting chondrogenic differen-tiation. (E) The expression of surface antigen markers for MSCs (CD29, CD90, CD44, and CD34) evaluated by flow cytometry assay. (F) Obser-vation of exosomes by TEM (scale bar: 200 nm). (G) Western blot analysis of the protein expression of exosome surface markers (CD63 and CD81), CANX, GM130, and Histone H3. MSC: mesenchymal stem cells, TEM: transmission electron microscopy, Exos: exosomes.
|
The results of TEM observation showed that the vesicles isolated from the supernatant of HUC-MSCs with or without transfection of miR-150-5p overexpression or inhibition lentiviral vectors by ultracentrifugation had a particle size of 50∼100 nm, which was accorded with the typical characteristics of exosomes; the transfection of the lentiviral vector exert no effect on the characteristics of exosomes (Fig. 1F). Western blot analysis showed the presence of the positive expression of exosome surface markers CD63 and CD81 but the absence of the expression of endoplasmic reticulum-derived CANX, GM130, and histone H3 in the vesicles isolated from the supernatant of the HUC-MSCs (Fig. 1G), suggesting the successful isolation of MSCs-derived exosomes.
MSCs-Exos promote the proliferation and migration of H2O2-induced HaCaT cells and inhibit the apoptosis
In order to study the functional effects of MSCs-Exos on skin injury, HaCaT cells were first induced with different concentrations of H2O2. According to the results of the CCK-8 assay, the viability of HaCaT cells decreased in a concentration-dependent manner (Fig. 2A). Meanwhile, the flow cytometry assay showed a concentration-dependent increase in the apoptosis rate (Fig. 2B). The results showed that a cellular model of skin injury was successfully established. When the concentration of H2O2 reached 300 μM, HaCaT cells showed lower viability and higher apoptosis rate, and thus 300 μM H2O2 was selected to treat the HaCaT cells for the follow-up experiments (the model group).
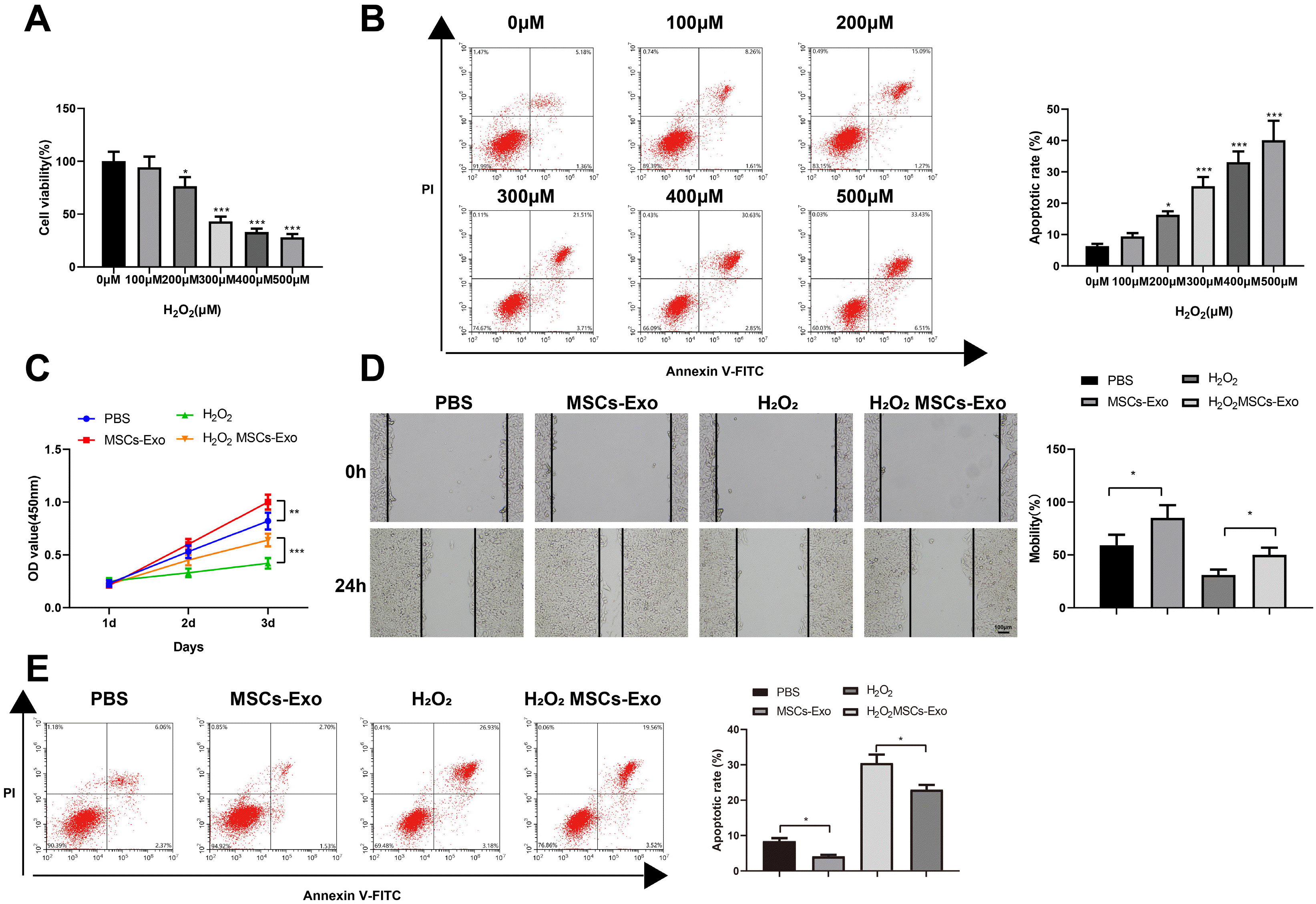 | Fig. 2MSCs-Exos promote the proliferation and migration and inhibit the apoptosis of H2O2-induced HaCaT cells. (A) Detection of the effects of H2O2 at different concentrations on the viability of HaCaT cells by CCK-8 method. (B) Detection of the apoptosis of HaCaT cells by flow cytometry. (C) Detection of the viability of HaCaT cells by the CCK-8 method. (D) Detection of the migration of HaCaT cells in each group by the plate scratch test. (E) Detection of the apoptosis of HaCaT cells by flow cytometry. *p<0.05, **p<0.01, ***p< 0.001, n=3. The data were presented in the form of mean±standard deviation. The comparison among multiple groups was performed by one-way analysis of variance. Tukey’s multiple tests were used for post hoc comparison. MSC: mesenchymal stem cell, Exos: exosomes.
|
Next, the proliferation, migration, and apoptosis of the HaCaT cells were tested. Compared with the PBS group, the OD450 value of the MSCs-Exo group increased. Compared with the H2O2 group, the OD450 value of the H2O2MSCs-Exo group increased. It was suggested that MSCs-Exo more effectively affected the proliferation of HaCaT cells injured by H2O2 (Fig. 2C, **p<0.01, ***p<0.001). After inhibiting the proliferation of HaCaT cells in each group using MMC, cell migration was detected using the plate scratch test. The results showed that the migration of HaCaT cells in the MSCs-Exo group was enhanced compared to the PBS group, and the migration of HaCaT cells in the H2O2MSCs-Exo group was promoted versus the H2O2 group, showing that MSCs-Exo promoted cell migration independent of proliferation (Fig. 2D, *p<0.05). The flow cytometry assay showed that the apoptosis of HaCaT cells in the MSCs-Exo group and the H2O2MSCs-Exo group was significantly inhibited (Fig. 2E, *p<0.05).
These results confirmed that the MSCs-Exos promoted the viability and migration and inhibit the apoptosis of HaCaT cells injured by H2O2.
Exosomal miR-150-5p effectively improves H2O2-induced injury in HaCaT cells
Previous studies have shown that miR-150-5p in small extracellular vesicles plays a role in promoting healing by increasing skin neovascularization and re-epithelialization at the tissue/cell level (15, 20). To study the effect of miR-150-5p expressed in the MSCs-Exos on H2O2-treated HaCaT cells, miR-150-5p expression was detected in HaCaT cells treated with different concentrations of H2O2. RT-qPCR showed that the level of miR-150-5p decreased with the increase of H2O2 concentration (Fig. 3A, *p< 0.05, ***p<0.001). At the same time, the expression of miR-150-5p in HUC-MSC culture medium supernatant after exosome isolation and exosome solution was detected by RT-qPCR, which showed that miR-150-5p was enriched in the exosome solution (Fig. 3B, ***p<0.001). In addition, miR-150-5p expression in extracellular vesicles was detected. RT-qPCR results showed that miR-150-5p expression did not change in the culture medium after RNase treatment. However, miR-150-5p expression decreased significantly after RNase and TritonX-100 treatment (Fig. 3C).
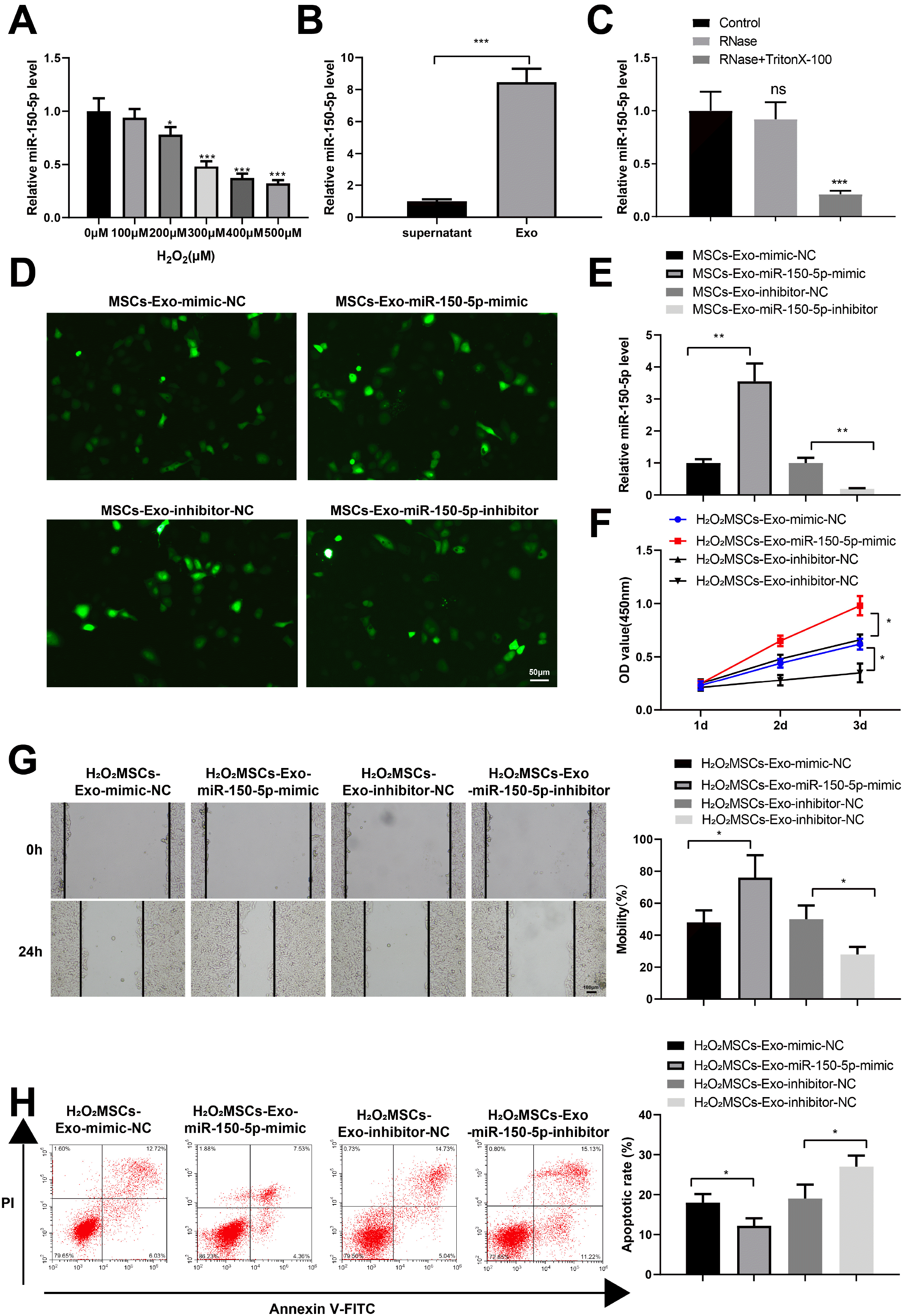 | Fig. 3MiR-150-5p expressed in the MSCs-Exos effectively improves H2O2-treated HaCaT cell viability. (A) Detection of miR-150-5p expression in HaCaT cells treated with H2O2 by RT-qPCR. (B) Detection of miR-150-5p expression in MSC culture medium supernatants after exosome isolation and exosomes solution by RT-qPCR. (C) Detection of miR-150-5p expression in exosomes after digestion with RNase by RT-qPCR. (D) Fluorescent images of lentivirus-infected cells. (E) Verification of the overexpression and inhibition efficacy of miR-150-5p by RT-qPCR. (F) Detection of the viability of HaCaT cells in each group by the CCK-8 method. (G) Determination of the migration of HaCaT cells in each group by the plate scratch test. (H) Detection of the apoptosis of HaCaT cells in each group by flow cytometry. *p<0.05, **p<0.01, ***p<0.001, n=3. The data were presented in the form of mean±standard deviation. The comparison among multiple groups was performed by one-way analysis of variance. Tukey’s multiple tests were used for the post hoc comparison. MSC: mesenchymal stem cells, Exos: exosomes.
|
To further investigate the effect of miR-150-5p expressed in the MSCs-Exos on H2O2-treated HaCaT cells, we infected the MSCs with miR-150-5p overexpression and inhibition lentiviral vectors and their NCs (Fig. 3D). Then MSCs-Exo-mimic-NC, MSCs-Exo-miR-150-5p-mimic, MSCs-Exo-inhibitor-NC, and MSCs-Exo-miR-150-5p-inhibitor were isolated and cultured with HaCaT cells treated with 300 μM H2O2. RT-qPCR results showed that miR-150-5p expression in the MSCs-Exo-miR-150-5p-mimic group was significantly enhanced than that in the MSCs-Exo-mimic-NC group; the MSCs-Exo-miR-150-5p-inhibitor group exhibited notably reduced miR-150-5p expression versus the MSCs-Exo-inhibitor-NC group (Fig. 3E, **p<0.01).
Compared with that in the H2O2MSCs-Exo-mimic-NC group, the OD450 value of the H2O2MSCs-Exo-miR-150-5p-mimic group was increased, which, however, was decreased in the H2O2MSCs-Exo-miR-150-5p-inhibitor group relative to the H2O2MSCs-Exo-inhibitor-NC group (Fig. 3F, *p<0.05). Cell migration after MMC treatment was detected using the plate scratch test. The results showed that HaCaT cells in the H2O2MSCs-Exo-miR-150-5p-mimic group showed remarkably higher migration ability compared with those in the H2O2MSCs-Exo-mimic-NC group; HaCaT cells in the H2O2MSCs-Exo-miR-150-5p-inhibitor group exhibited notably reduced migration ability relative to those in the H2O2MSCs-Exo-inhibitor-NC group (Fig. 3G, *p<0.05). According to the flow cytometry assay results, compared with that in the H2O2MSCs-Exo-mimic-NC group, the apoptosis of HaCaT cells in the H2O2 MSCs-Exo-miR-150-5p-mimic group was significantly inhibited; the H2O2MSCs-Exo-miR-150-5p-inhibitor group exhibited the dramatically elevated apoptosis rate of HaCaT cells compared with the H2O2MSCs-Exo-inhibitor-NC group (Fig. 3H, *p<0.05). These results suggested that exosomal miR-150-5p from MSCs promoted the proliferation and migration and inhibited the apoptosis of H2O2-injured HaCaT cells.
MiR-150-5p regulates PI3K/AKT pathway via PTEN
The starBase tool (http://starbase.sysu.edu.cn/) predicted that miR-150-5p might target PTEN (Fig. 4A). In addition, PTEN expression was detected by RT-qPCR and western blot analyses in HaCaT cells treated with different concentrations of H2O2, which reported that PTEN level increased in a concentration-dependent manner (Fig. 4B and 4C, *p<0.05, **p<0.01, ***p<0.001). Moreover, by detecting the luciferase activities, we found that miR-150-5p mimic decreased the luciferase activity of HEK-293T cells transfected with WT-PTEN (**p<0.01), but had no effect on the luciferase activity of HEK-293T cells transfected with MUT-PTEN (Fig. 4D, p>0.05). RIP assay further confirmed the interaction between miR-150-5p and PTEN (Fig. 4E). Moreover, the expression of miR-150-5p and PTEN and the phosphorylation of PI3K and AKT were detected by RT-qPCR and western blot analysis in HaCaT cells transfected with miR-150-5p mimic, miR-150-5p inhibitor, or miR-150-5p inhibitor+sh-PTEN. The results showed that compared with the mimic NC group, the expression levels of miR-150-5p, p-PI3K, and p-AKT in the miR-150-5p mimic group increased significantly, while the PTEN mRNA and protein expression decreased significantly (Fig. 4F and 4G, **p<0.01, ***p<0.001); the expression levels of miR-150-5p, p-PI3K and p-AKT substantially decreased, and the expression levels of PTEN mRNA and protein increased in the miR-150-5p inhibitor group, compared with the inhibitor NC group (Fig. 4F and 4G, **p<0.01, ***p<0.001). Compared with the miR-150-5p inhibitor group, the expression of p-PI3K and p-AKT in the miR-150-5p inhibitor+sh-PTEN group increased significantly, while the expression of PTEN mRNA and protein decreased substantially. These results suggest that miR-150-5p regulates PI3K/AKT pathway by regulating PTEN.
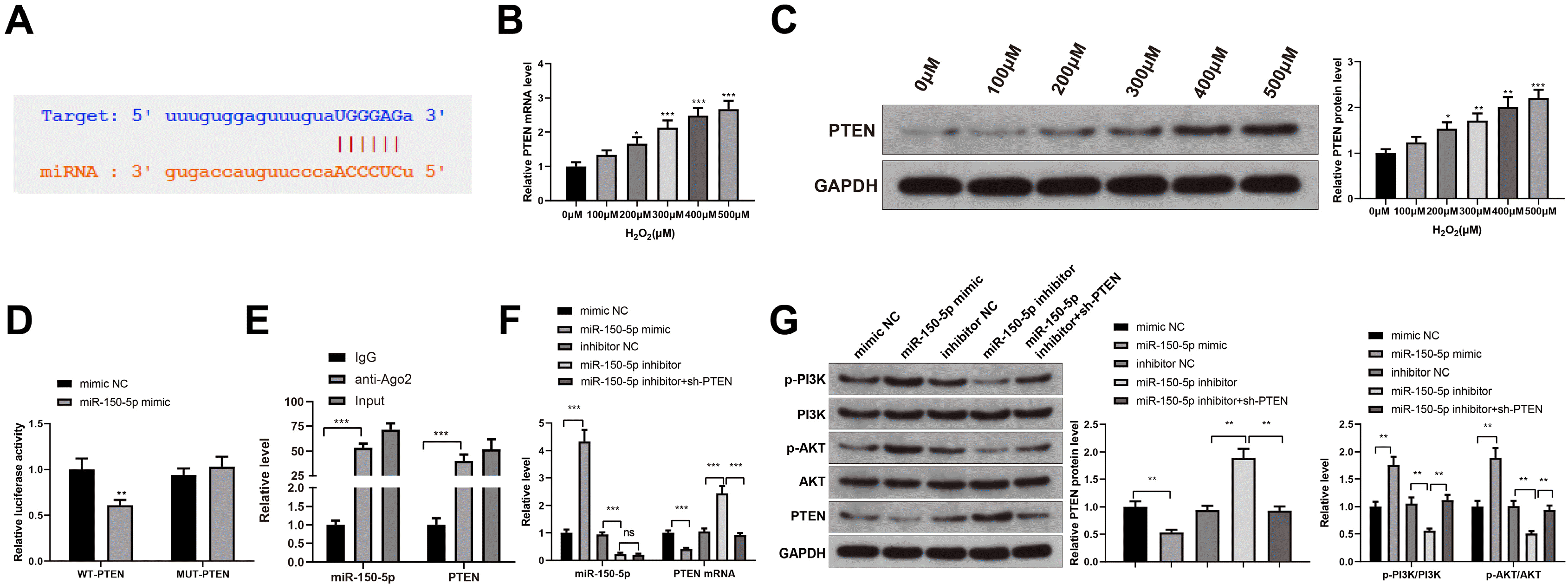 | Fig. 4MiR-150-5p regulates PI3K/AKT pathway by mediating PTEN. (A) Prediction of the binding site between miR-150-5p and PTEN by starBase (http://starbase.sysu.edu.cn/). (B, C) The detection of the expression level of PTEN in HaCaT cells treated with different concentrations of H2O2 by RT-qPCR (B) and western blot analysis (C). (D, E) Verification of the interaction between miR-150-5p and PTEN by dual-luciferase reporter assay (D) and RIP test (E). (F) The detections of miR-150-5p expression and PTEN mRNA expression in HaCaT cells transfected with miR-150-5p mimic, miR-150-5p inhibitor, or miR-150-5p inhibitor+sh-PTEN by RT-qPCR. (G) PTEN, PI3K, and AKT protein expression and the phosphorylation of PI3K and AKT in HaCaT cells transfected with miR-150-5p mimic, miR-150-5p inhibitor, or miR-150-5p inhibitor+sh-PTEN by western blot analysis. *p<0.05, **p<0.01, ***p<0.001, n=3. The data were presented in the form of mean±standard deviation. The comparison among multiple groups was performed by one-way analysis of variance. Tukey’s multiple tests were used for the post hoc comparison. MSC: mesenchymal stem cells, Exos: exosomes.
|
PTEN overexpression counteracts the protective effect of exosomal miR-150-5p on H2O2-induced HaCaT cells
To further study the role of PTEN in the exosomal miR-150-5p-mediated inhibition in the cellular model of skin injury, HaCaT cells transfected with pcDNA3.1 or pcDNA3.1-PTEN were co-cultured with MSCs-Exo-NC or MSCs-Exo-miR-150-5p. The transfection efficacy of pcDNA3.1-PTEN was confirmed by RT-qPCR and western blot analyses (Fig. 5A and 5B). Following incubation with 300 μM H2O2-treated HaCaT cells, RT-qPCR and western blot analyses were used to detect PTEN expression in H2O2-treated HaCaT cells transfected with pcDNA3.1 or pcDNA3.1-PTEN (Fig. 5C). The results revealed that compared with that in the H2O2MSCs-Exo-NC+NC group, PTEN mRNA and protein levels were notably decreased in the H2O2MSCs-Exo-miR-150-5p+NC group and dramatically increased in the H2O2MSCs-Exo-NC+PTEN group; relative to the H2O2MSCs-Exo-miR-150-5p+ NC group, the H2O2MSCs-Exo-miR-150-5p+PTEN group exhibited increased PTEN expression (Fig. 5C and 5D).
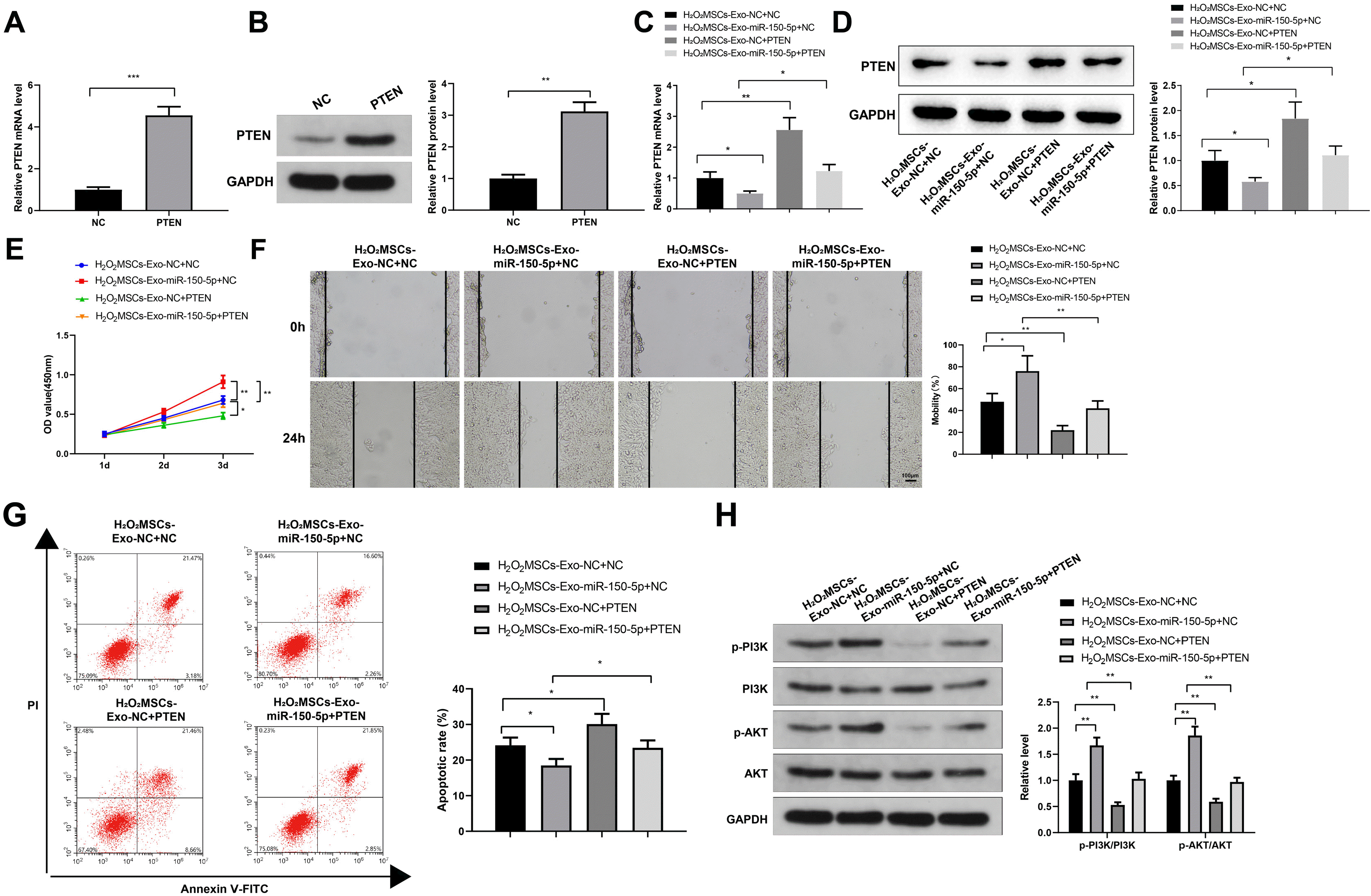 | Fig. 5PTEN overexpression restrains exosomal miR-150-5p-mediated inhibition on H2O2-induced injury in HaCaT cells. (A, B) The expression of PTEN in HaCaT cells transfected with pcDNA3.1 or pcDNA3.1-PTEN by RT-qPCR (A) and western blot analysis (B). (C, D) The expression of PTEN in HaCaT cells by RT-qPCR (C) and western blot analysis (D). (E) Detection of the viability of HaCaT cells by the CCK-8 method. (F) Detection of the migration of HaCaT cells in each group by the plate scratch test. (G) Detection of apoptosis of HaCaT cells in each group by flow cytometry. (H) Detection of the phosphorylation level of PI3K and AKT by western blot analysis. *p<0.05, **p<0.01, ***p<0.001, n=3. The data were presented in the form of mean±standard deviation. The comparison among multiple groups was performed by one-way analysis of variance. Tukey’s multiple tests were used for the post hoc comparison. MSC: mesenchymal stem cells, Exos: exosomes.
|
Compared with the H2O2MSCs-Exo-NC+NC group, the OD450 value increased in the H2O2MSCs-Exo-miR-150-5p+NC group, while decreased in the H2O2MSCs-Exo-NC+PTEN group; and the OD450 value of the H2O2MSCs-Exo-miR-150-5p+PTEN group decreased compared with the H2O2MSCs-Exo-miR-150-5p+NC group (Fig. 5E, *p< 0.05, **p<0.01).
Cell migration after MMC treatment was detected using the plate scratch test. The results showed that the H2O2MSCs-Exo-miR-150-5p+NC group showed enhanced migration ability, while the H2O2MSCs-Exo-NC+PTEN group presented weakened migration ability, compared with the H2O2MSCs-Exo-NC+NC group; the H2O2MSCs-Exo-miR-150-5p+PTEN group exhibited reduced migration ability relative to the H2O2MSCs-Exo-miR-150-5p+NC group (Fig. 5F, *p<0.05, **p<0.01).
According to the results of flow cytometry, the apoptosis was decreased in the H2O2MSCs-Exo-miR-150-5p+NC group, and increased in the H2O2MSCs-Exo-NC+PTEN group, compared with the H2O2MSCs-Exo-NC+NC group; the apoptosis rate of the H2O2MSCs-Exo-miR-150-5p+PTEN group was higher than that of the H2O2MSCs-Exo-miR-150-5p+NC group (Fig. 5G, *p<0.05). These results suggested that PTEN attenuated the protective effect of miR-150-5p expressed in the MSCs-Exos on H2O2-induced HaCaT cells.
Western blot analysis was used to detect the phosphorylation level of PI3K and AKT. Compared to the H2O2 MSCs-Exo-NC+NC group, the expression of p-PI3K and p-AKT increased in the H2O2MSCs-Exo-miR-150-5p+NC group, while decreasing in the H2O2MSCs-Exo-NC+PTEN group. Also, the expression levels of p-PI3K and p-AKT in the H2O2MSCs-Exo-miR-150-5p+PTEN group were lower than that in the H2O2MSCs-Exo-miR-150-5p+NC group (Fig. 5H, **p<0.01).
Go to :
Discussion
The process of wound healing includes hemostasis, inflammation, hyperplasia, and remodeling (21). Delayed wound healing greatly increases the risk of infection. In this study, for the first time, our in vitro experimental results provide a qualitative and quantitative basis for MSCs-derived miR-150-5p-expressing exosomes provoking keratinocyte proliferation and migration by activating PI3K/AKT pathway through PTEN. This intermolecular association, which exists but has never been studied in-depth, may contribute to evoking the regenerative responses, thereby accelerating skin wound repair and regeneration.
Initially, the MSCs-Exos were found to promote the proliferation and migration and inhibit the apoptosis of H2O2-induced HaCaT cells effectively. Increasing evidence showed that the main mechanism of stem cell transplantation therapy may fundamentally depend on the paracrine activity, that is, stem cells secrete bioactive molecules that impact the biological functions of neighboring cells (22, 23). The direct use of exosomes may overcome the limitations and risks associated with stem cell transplantation (1). A considerable number of studies proved that MSCs-Exos promote wound healing depending on the bioactive cargoes they load. For instance, HOTAIR-loaded MSCs-derived extracellular vesicles contribute to angiogenesis and wound healing (24). miR-31-5p-expressing exosomes were found, through in vitro and in vivo experiments, to enhance diabetic wound healing by enhancing angiogenesis, fibrogenesis, and re-epithelization (25).
Next, we narrowed down the main range of the independent variable to find its effective components and found that MSCs-derived exosomal miR-150-5p could improve the viability and migration of H2O2-induced HaCaT cells, which led to speculation that miR-150-5p might be a key player in exosome-accelerated wound healing. The promoting effect of miR-150-5p on cancer cell growth and migration has been identified in non-small cell lung cancer and colon cancer (18, 26). Interestingly, MSC-derived miR-150-5p-expressing exosomes were found to decrease the migration and invasion of fibroblast-like synoviocytes and reduce the tube formation of human umbilical vein endothelial cells in rheumatoid arthritis (27). Wu et al. (28) demonstrated that bone marrow MSC-derived exosomal miR-150-5p suppressed the apoptosis of cardio-myocytes induced by myocardial infarction. The present study adds to the preexisting knowledge on the role of exosomal miR-150-5p in keratinocyte proliferation and migration.
In addition, bioinformatics prediction result using the online site starBase (miR-150-5p targeting PTEN) provides us with a way forward, and then the results of RT-qPCR and western blot analyses revealed the association between miR-150-5p and PTEN expression, and the phosphorylation of PI3K and AKT, suggesting a direct or indirect cause and effect relationship between them. PTEN is related to the downregulation of the PI3K/AKT pathway and acts as an anti-tumor gene by mediating the cell cycle, apoptosis, and migration (29). PTEN inhibition has been demonstrated to accelerate wound healing in corneal endothelia by increasing the division and migration of endothelial cells (30). MiR-21-mediated inhibition of PTEN activated the PI3K/AKT pathway and contributed to skin wound healing (31). At the same time, studies have shown that activating the PI3K/AKT pathway can accelerate skin wound healing (32, 33). Finally, we use the method of callback, taking PTEN as the variable, from three aspects of cell proliferation, migration, and apoptosis to further verify the scientific results of the experiment, combined with the phosphorylation evaluations of PI3K and AKT. The results showed that PTEN overexpression counteracted the protective effect of exosomal miR-150-5p on H2O2-induced HaCaT cells.
In summary, our results show that MSCs-derived miR-150-5p-expressing exosomes promote skin wound healing by activating PI3K/AKT pathway through mediating PTEN expression. Despite that the pro-healing effect of exosomal miR-150-5p was proven at a cellular level, the interpretation of this study should be cautious since the present work investigated little on the skin injury at the animal level. Our findings suggest MSCs-Exos as a novel and effective therapeutic tool for improving skin/soft tissue wound healing in the future.
Go to :
 XML Download
XML Download