

 PDF
PDF Citation
Citation Print
Print



INTRODUCTION
Non-alcoholic fatty liver disease (NAFLD) is the most common chronic liver disease with a worldwide prevalence of 20% to 30% [1], and its prevalence is even higher in high caloric intake and obese populations [2]. Though NAFLD is commonly benign, it may develop into inflammation, fibrosis, cirrhosis (non-alcoholic steatohepatitis [NASH]), and eventually cancer of the liver [3]. NAFLD is a metabolic disorder caused by the accumulation of fat in the liver leading to liver dysfunction [4]. Unfortunately, specific and potent treatment options for NAFLD have not been recognized yet. Thus, there is an urgent need for exploring strategies for the proper treatment of NAFLD.
A number of studies have reported a pathogenic role of oxidative stress in NAFLD [5]. Markers of oxidative stress such as lipid peroxidation and reactive oxygen species (ROS) are increased in the liver of NAFLD patients [6]. The accumulation of triglycerides (TG) in the cytoplasm of hepatocytes is associated with NAFLD, and exposure to hydrogen peroxide (H2O2) increases the cytoplasmic TG level in HepG2 cells, a human hepatocyte carcinoma cell line [7]. Besides, various antioxidants, including curcumin, resveratrol, quercetin, and lycopene, decrease oxidative stress and the features of NAFLD [8]. Thus, inhibition of oxidative stress may play a key role in inhibiting NAFLD progression.
Peroxisomes are single membrane-bound organelles that rapidly assemble, multiply, and are degraded in response to metabolic needs. Peroxisomal biogenesis is regulated by de novo biogenesis, the growth and division of pre-existing peroxisomes, and pexophagy [9]. de novo biogenesis requires the fusion of two pre-peroxisomal vesicles, one from the endoplasmic reticulum (ER) and the other from mitochondria [10]. Proteins that are involved in the peroxisomal biogenesis process are called peroxins (peroxisomal biogenesis factor [PEX]). Peroxisomes regulate many metabolic functions, such as the β-oxidation of fatty acids (FA) as well as ROS homeostasis [11]. Peroxisomal β-oxidation of very-long-chain fatty acids (VLCFA) occurs through ATP binding cassette subfamily D member 1 (ABCD1), while α-oxidation of branched-chain fatty acids (BCFA), synthetic ether-chain phospholipids and bile acids occurs through ABCD3 (also named as PMP70) [12]. Pex2 deficiency increases cholesterol synthesis in the liver of newborn mice [13]. Pex11b deficiency increases neuronal apoptosis and causes defects in peroxisomal FA β-oxidation and peroxisomal ether lipid biosynthesis in Zellweger syndrome mice [14]. Also, Pex11b deficiency results in developmental delay of kidneys and livers [14].
The peroxisome has a dense crystalline core within a large number of antioxidant enzymes, such as catalase, peroxiredoxin (PRX) 1, 5, and 6, copper- and zinc-containing superoxide dismutase (Cu/ZnSOD), and epoxide hydrolase, which may play important roles in ROS metabolism [12]. Endogenous catalase has protective effects on the kidney from diabetic stress through maintaining peroxisomal fitness [15]. Also, redox imbalance in peroxisomes of catalase knockout mice accelerates NAFLD in mice [16]. Impaired peroxisomal fitness may enhance oxidative stress and inflammation in white adipose tissue leading to obesity [17]. Thus, any alteration in peroxisomal function can potentially exacerbate the oxidative stress leading to tissue injury [15-18]. Accordingly, the loss of peroxisomes and impaired peroxisomal functions have been demonstrated to occur in inflammatory conditions, including NASH and NAFLD [16,18].
Fenofibrate 2-[4-(4-chlorobenzoyl)phenoxy]-2-methylpropanoic acid, 1-methylethyl ester), a peroxisome proliferator-activated receptor α (PPARα) agonist, is widely used in the clinic as a lipid-lowering agent against mixed dyslipidemia or primary hypercholesterolemia [19], while PPARα is the key regulator of fatty acid oxidation (FAO) [20]. Fenofibrate reduces the activity of acetyl coenzyme A carboxylase (ACC) and fatty acid synthase (FAS), thus inhibiting the de novo synthesis of FAs. Fenofibrate may reduce the risk of cardiovascular disease and diabetic retinopathy in diabetic patients [21,22]. Fenofibrate protects mice against high-fat diet (HFD)-induced kidney injury [23]. Also, fenofibrate induces the expression of Pex11a in the kidney, which may increase the elongation and number of peroxisomes [24]. Fenofibrate prevents fasting-refeeding process-induced abnormal liver function by increasing peroxisomal FAO and peroxisome biogenesis [25]. On the other hand, a PPARα independent action of fenofibrate also has been reported in various tissues and cells [23,26,27]. Thus, it is important to understand the underlying molecular mechanisms of fenofibrate-mediated peroxisomal fitness in the liver.
The purpose of this study was (1) to evaluate the role of the peroxisome and (2) to determine the involvement of PPARα in fenofibrate-induced peroxisomal fitness against HFD-induced NAFLD in mice.
METHODS
Materials
Chemicals and immunoblotting antibodies were obtained from Sigma-Aldrich Company (St. Louis, MO, USA), Nunc (Rochester, NY, USA), and Cell Signaling Technology (Danvers, MA, USA), respectively, unless otherwise stated.
Animals
In series I, 8-week-old male C57BL/6J wild-type (WT) mice were used. In series II, 8-week-old male Ppara-deficient (Ppara-/-) [28] mice were used. The mice were housed in a temperature-controlled room on a 12-hour light-dark cycle. They were fed a normal diet (ND; 10% fat, Research Diets D12450) or a HFD (60% fat, Research Diets D12492) for 12 weeks [17]. Fenofibrate (F6020, Sigma-Aldrich) was prepared at 0.5% dissolved in carboxymethyl cellulose (CMC) and administered to ND, HFD, and HFD Ppara-/- mice daily at a dose of 50 mg/kg (200 to 400 μL/mice) by oral gavage for 12 weeks. ND and HFD mice not treated by fenofibrate were administered with an equal volume of CMC. After 12 weeks, all mice were sacrificed. Blood plasma and liver tissues were collected for further analysis. All animal studies were approved by the Institutional Animal Care and Use Committee of Ewha Womans University (No.15-062 and No.18-054).
Measurement of blood parameters
Blood samples were collected with a heparinized syringe and centrifuged at 3,000 rpm for 20 minutes at 4°C to collect the plasma. Plasma alanine aminotransferase (ALT) levels were measured using an EnzyChrom L-Alanine Assay Kit (EALA-100, BioAssay Systems, Hayward, CA, USA).
Real-time polymerase chain reaction
Tissue samples were subjected to real-time polymerase chain reaction (qPCR) analysis as previously described [15]. Briefly, the mRNA levels were measured by qPCR using a SYBR Green PCR Master Mix kit (Applied Biosystems, Foster City, CA, USA) with the StepOne Real-Time PCR System (Applied Biosystems). The relative quantitation of each gene was calculated after normalization to 18S rRNA levels. The primer sequences are listed in Supplementary Table 1.
Histology and immunohistochemistry staining
Tissue samples were subjected to immunohistochemistry (IHC) analysis as previously described [15]. Briefly, liver tissue was fixed in 4% paraformaldehyde-lysine-periodate, dehydrated, and embedded in paraffin. To examine the liver histology and morphology, 5 μm liver sections were stained with hematoxylin and eosin (H&E). For IHC staining, anti-4-hydroxynonena (4-HNE; 1:200, MHN-100P, CiteAb, New Bond St, UK), anti-F4/80 (1:200; Santa Cruz Biotechnology Inc., Dallas, TX, USA), anti-nitrotyrosine (NT, 1:400, sc-32757, Santa Cruz Biotechnology Inc.), anti-8-hydroxyguanine (8-oxo-dG; 1:200; 4354-MC-050, Trevigen, Gaithersburg, MD, USA), and anti-collagen 1 (COL1; 1:200, 1310-01, Southern Biotech, Birmingham, AL, USA) antibodies were used and incubated with the tissue sections overnight at 4°C. Images were captured using a Zeiss microscope equipped with an Axio Cam HRC digital camera and Axio Cam software (Carl Zeiss, Thornwood, NY, USA). The staining intensities were quantified using Image-Pro Plus 4.5 software (Cybernetics, Silver Spring, MD, USA).
Immunofluorescence staining
Liver sections were incubated with the indicated primary antibodies, such as anti-ABCD3 (1:200, ab85550, Abcam, Cambridge, UK), anti-catalase (1:200, sc271803, Santa Cruz Biotechnology Inc.), and anti-adipose differentiation-related protein (ADFP, 1:200, ab52356, Abcam). After incubation with the primary antibodies, the liver sections were subsequently incubated with Alexa 488-conjugated goat anti-mouse (1:1,000, A11018, Invitrogen, Carlsbad, CA, USA) and Alexa 568-conjugated goat anti-rabbit (1:1,000, A11070, Invitrogen). 4´,6-Diamidino-2-phenylindole dihydrochloride (DAPI, 1:1,000, 62248, Thermo Fisher Scientific, Waltham, MA, USA) was used for cell nuclei staining.
Immunoblotting analysis
Tissue samples were subjected to immunoblotting analysis as described previously [15]. Briefly, liver tissue was homogenized in lysis buffer and centrifuged at 13,000 rpm, 4°C for 15 minutes. The total concentration of protein was measured using the Bradford protein assay dye (Bio-Rad Laboratories, Hercules, CA, USA). The whole lysate was mixed with 5× sample buffer and denatured at 95°C for 6 minutes. The total proteins were separated by SDS-PAGE gel electrophoresis and transferred onto a polyvinylidene fluoride membrane (GE Healthcare BioSciences Co., Piscataway, NJ, USA). After protein blocking, the membranes were incubated with anti-phospho-nuclear factor kappa B (p-NF-κB, 1:1,000, #3033, Cell Signaling Technology), anti-total-nuclear factor kappa B (t-NF-κB, 1:1,000, #8243, Cell Signaling Technology), anti-acyl-CoA oxidase 1 (ACOX1; 1:1,000, sc-98499, Santa Cruz Biotechnology Inc.), and anti-β-actin (1:3,000, A5326, Sigma-Aldrich) antibodies. The blots were reacted with peroxidase-conjugated secondary antibodies (Vector Laboratories Inc., Burlingame, CA, USA), and the positive immunoreactive protein bands were detected using LAS-3000 film (FUJIFILM Corporation, Tokyo, Japan). All protein levels were normalized to β-actin.
Measurement of peroxisomal FAO
Liver FAO levels were measured as previously described [17]. Liver tissue (50 mg) was placed in reaction buffer containing 0.2 mM palmitate (NEC075H250UC, 14C-palmitate at 1.25 μCi/mL, NEN Life Science, Boston, MA, USA). Homogenized liver samples were incubated on an orbital shaker-incubator (Vision, Daejeon, Korea) at 30°C. The FAO reaction produced 14CO2 was trapped with 1 N NaOH solution. After 2 hours of incubation, the reaction was stopped by the addition of 4 N sulfuric acid. The trapped 14CO2 solution was mixed with a liquid scintillation cocktail (Ultima Gold, PerkinElmer, Waltham, MA, USA) and measured using a Packard TopCount NXT Luminescence and Scintillation Counter (Packard, San Diego, CA, USA). Peroxisomal FAO was measured in the reaction buffer in the presence of 100 µM antimycin A and 12.5 µM rotenone.
RESULTS
Fenofibrate ameliorates HFD-induced liver injury in mice
HFD feeding for 12 weeks accelerated the gain of body weight, and body weight at the time of sacrifice were 25.5±0.5 and 33.7±0.5 g in ND- and HFD-fed mice (P<0.05), respectively. Fenofibrate effectively prevented HFD-induced weight gain, and body weight of treated HFD mice were 30.1±0.4 g (P<0.05 compared to HFD-fed mice). The protective effects of fenofibrate against HFD-induced liver injury have been established [25,29,30]. Consistently, immunofluorescence (IF) staining of ADFP (also called as perilipin-2), a marker of lipid droplets, was increased in HFD-fed mice liver and inhibited by fenofibrate (Fig. 1A) in the present study. Macrophage infiltration in the fatty liver was elevated as indicated by an increase in F4/80-positive staining area, which was significantly decreased by fenofibrate treatment (Fig. 1A and B). 8-oxo-dG, NT, and 4-HNE immunostaining were used to determine the state of oxidative stress in HFD mice after fenofibrate treatment. HFD significantly increased 8-oxo-dG, NT, and 4-HNE accumulation, which were inhibited by fenofibrate (Fig. 1A, C, D, and E). HFD-induced Il1b, Il6, F4/80, and monocyte chemoattractant protein 1 (Mcp1) mRNA levels were also inhibited by fenofibrate (Fig. 1F). Also, the protein levels of liver p-NF-κB were increased in HFD-fed mice, which were inhibited by fenofibrate (Fig. 1G and H). HFD-fed mice showed significantly increased plasma ALT levels, which were effectively decreased by fenofibrate (Fig. 1I).
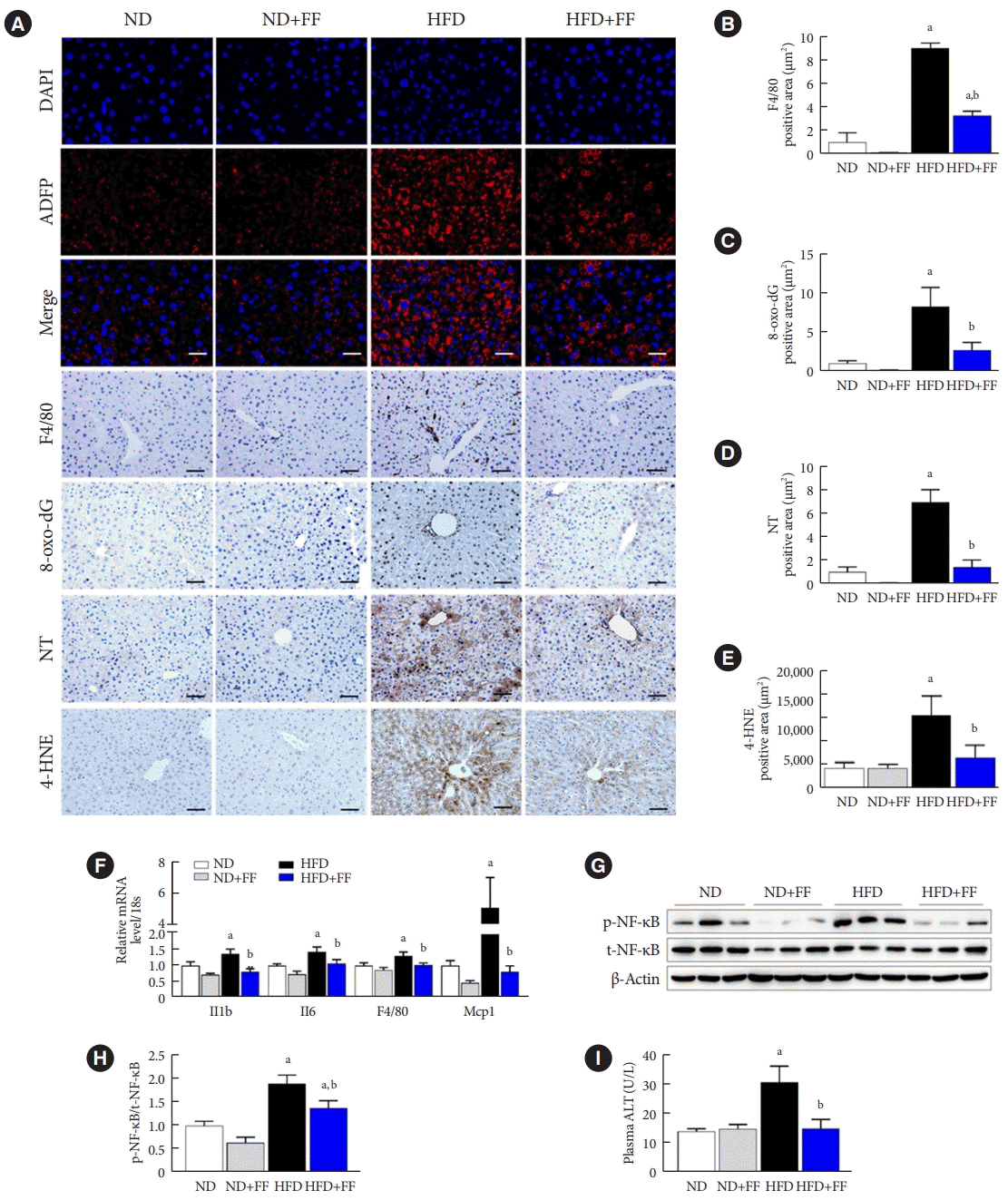
Fig. 1.
Fenofibrate (FF) ameliorates high-fat diet (HFD)-induced liver dysfunction in wild-type (WT) mice. (A) Liver sections were immunofluorescence (IF) stained for adipose differentiation-related protein (ADFP; red) and 4´,6-diamidino-2-phenylindole dihydrochloride (DAPI) nuclear counterstaining (blue). Original magnification, 200×; scale bar, 100 μm. (A, B, C, D, E) Liver sections were also stained with anti-F4/80, 8-hydroxyguanine (8-oxo-dG), nitrotyrosine (NT), and 4-hydroxynonena (4-HNE) antibodies and were quantified. Original magnification, 100×; scale bar, 200 μm, n=4. (F) Interleukin 1β (Il1b), Il6, F4/80, and monocyte chemoattractant protein 1 (Mcp1) were measured by real-time polymerase chain reaction, and the results were normalized to the 18S rRNA levels. (G, H) The protein levels of phospho-nuclear factor kappa B (p-NF-κB) and total-NF-κB (t-NF-κB) were measured by Western blotting. (I) Plasma alanine aminotransferase (ALT) levels were measured using an enzyme-linked immunosorbent assay (ELISA) kit. Data are expressed as the mean±standard error of 6 mice/group. ND, normal diet. aP<0.05 vs. ND mice, bP<0.05 vs. HFD mice.
![]()
Fenofibrate increases liver peroxisomal biogenesis in HFD mice
HFD impairs liver peroxisomal biogenesis, resulting fatty liver in mice [16]. Fenofibrate restores fasting/refeeding-induced impairment of liver peroxisomal biogenesis in mice [25]. Interestingly, in the current study, expression of Pex3, Pex5, Pex13, Abcd1, and Acox1 mRNA was significantly decreased in HFD-fed mice liver (Fig. 2A). And nine of 13 analyzed genes (Pex5, Pex7, Pex11, Pex13, Pex16, Pex19, Abcd2, Abcd3, and Acox1) involved in peroxisomal fitness were upregulated in the liver of fenofibrate-treated HFD mice compared to each of HFD-fed mice. The administration of fenofibrate did not affect the expression of any gene in ND-fed mice. We, thus, measured mRNA expression of PPARα target genes such as cluster of differentiation 36 (Cd36), fatty-acid-binding protein (Fabp), and peroxisome proliferator-activated receptor co-activator-1α (Pgc1a). Fenofibrate upregulated the expression of Cd36 and Fabp, but not Pgc1a, mRNA in ND-fed mice (Supplementary Fig. 1). These data suggest that not all target genes are simultaneously regulated to the same extent.
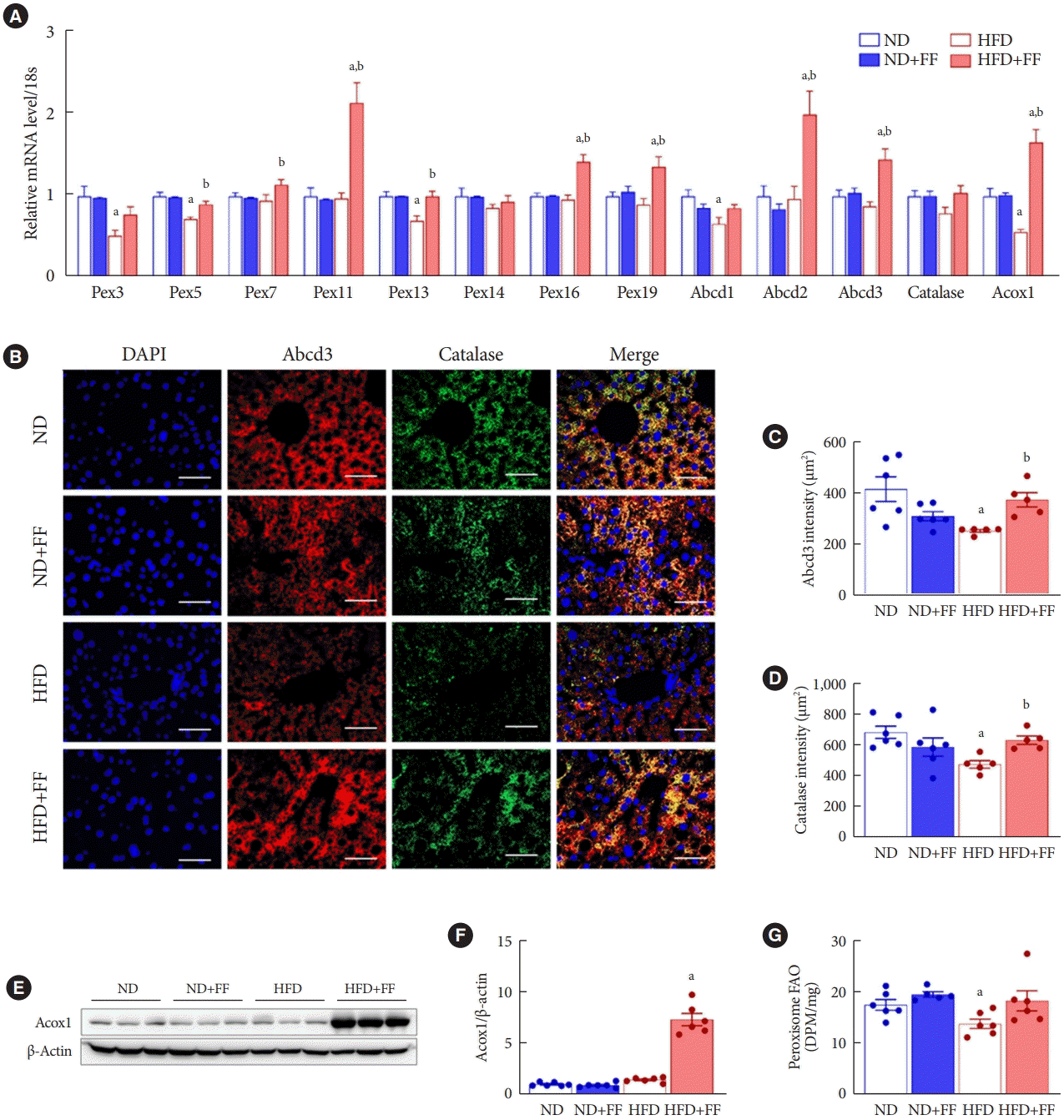
Fig. 2.
Fenofibrate (FF) improves liver peroxisomal function in high-fat diet (HFD)-fed wild-type (WT) mice. (A) Peroxisome-related genes were analyzed by real-time polymerase chain reaction, and the results were normalized to 18S rRNA levels. (B, C, D) Liver sections were used for immunofluorescence (IF) staining of ATP binding cassette subfamily D member 3 (ABCD3; red), catalase (green), and 4´,6-diamidino-2-phenylindole dihydrochloride (DAPI) counterstaining (blue) and were quantified. Original magnification, 200×; scale bar, 200 μm. (E, F) Protein levels of acyl-CoA oxidase 1 (ACOX1) were measured by Western blotting, and the results were normalized to β-actin levels. (G) Peroxisomal fatty acid oxidation (FAO) was measured in liver tissue. Data are expressed as the mean±standard error of 6 mice/group. ND, normal diet; Pex, peroxisomal biogenesis factor; DPM, disintegration per minute. aP<0.05 vs. ND mice, bP<0.05 vs. HFD mice.
![]()
Fenofibrate improves liver peroxisomal function in HFD mice
Catalase is the most abundant peroxisomal antioxidant enzyme [31], while ABCD3, a major component of the peroxisomal membrane, is involved in metabolic transport of long-chain acyl-CoA [32]. To examine the effect of fenofibrate on peroxisomal function, we co-stained ABCD3 and catalase in the liver sections. HFD-fed mice showed decreased expression of ABCD3 and catalase, which were effectively inhibited by fenofibrate (Fig. 2B-D), suggesting that fenofibrate increased functional peroxisomes in the fatty liver. Fenofibrate increased the protein levels of ACOX1, a rate-limiting enzyme in the peroxisomal β-oxidation pathway, in HFD-fed mice without significant effect on ND-fed mice (Fig. 2E and F). Importantly, HFD feeding significantly reduced peroxisomal FAO in mice liver, which was also effectively increased by fenofibrate (Fig. 2G).
PPARα is important for maintaining liver homeostasis in mice
PPARα activation is implicated in improving steatosis, inflammation, and fibrosis in various pre-clinical models of NAFLD [33]. Hepatocyte-specific deletion of Ppara promotes NAFLD phenotypes even under ND in mice [34]. In silico analysis using Gene Expression Omnibus (GES83452) data of human liver biopsy of normal and NASH showed decreased PPARA expression in patients with NASH (Supplementary Fig. 2). Interestingly, increased lipid accumulation in Ppara-/- mice under ND was exacerbated by HFD (Fig. 3A). Deficiency of Ppara increased the levels of 4-HNE and COL1 even under ND, and HFD feeding further increased COL1 immunostaining in Ppara-/- mice (Fig. 3A-C). Basal expression of Il1b, Il6, and F4/80 mRNA were significantly increased in Ppara-/- mice (Fig. 3D-F). Consistently, a deficiency of Ppara increased plasma ALT levels in mice even under ND (Fig. 3G). Fenofibrate failed to reduce HFD-induced ALT in Ppara-/- mice (Supplementary Fig. 3).
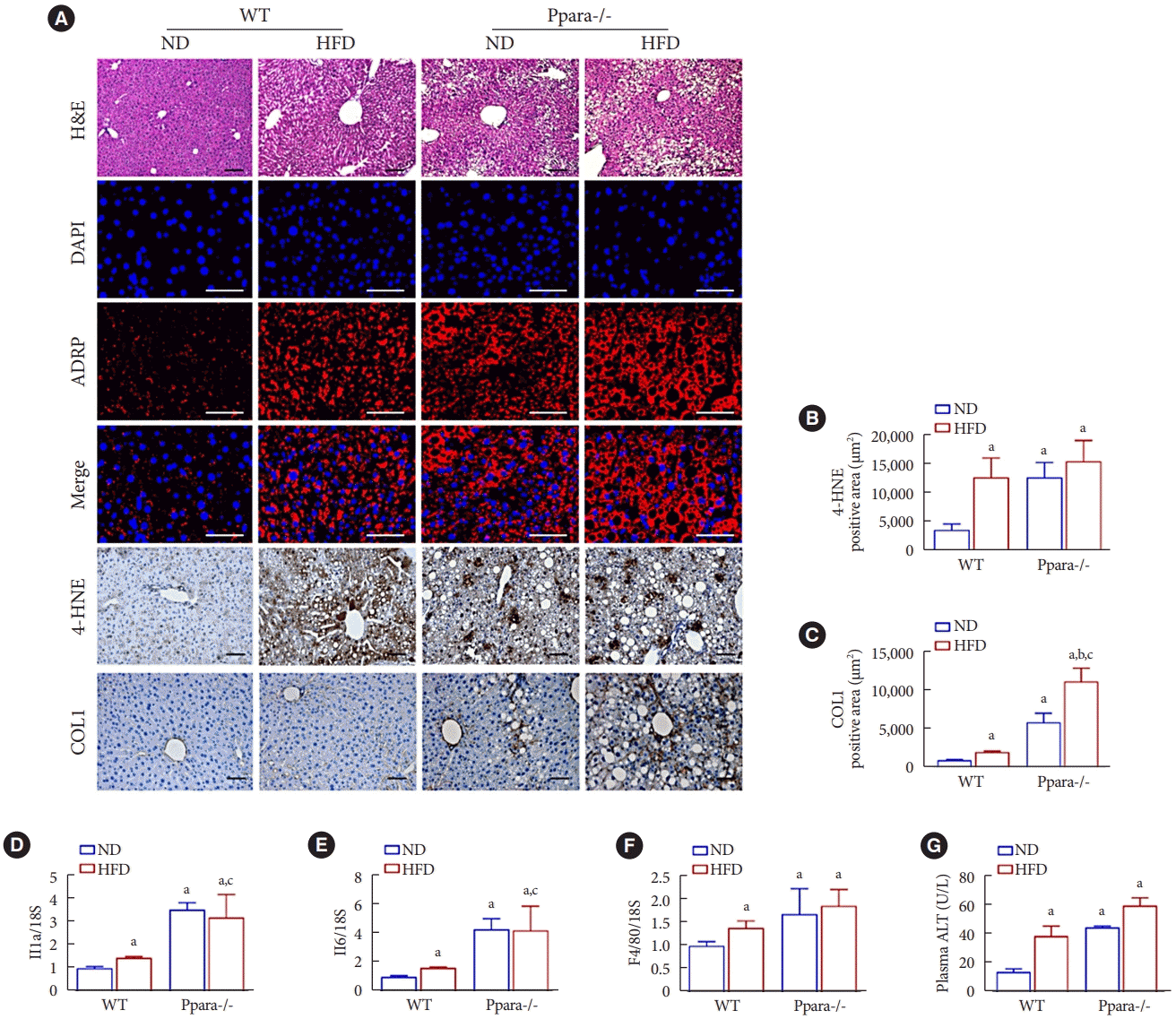
Fig. 3.
Role of peroxisome proliferator-activated receptor α (PPARα) in maintaining liver homeostasis in Ppara-/- mice. (A, B, C) Liver morphology was detected by H&E staining. Original magnification, 100×; scale bar, 200 μm. Liver sections were immunofluorescence (IF) stained for adipose differentiation-related protein (ADFP; red) and 4´,6-diamidino-2-phenylindole dihydrochloride (DAPI) nuclear counterstaining (blue). Original magnification, 200×; scale bar, 200 μm. Liver sections were also stained with anti-4-hydroxynonena (4-HNE) and collagen 1 (COL1) antibody and the positive area were quantified. Original magnification, 100×; scale bar, 200 μm. (D, E, F) Interleukin 1β (Il1b), Il6, and F4/80 were measured by real-time polymerase chain reaction, and the results were normalized to the 18S rRNA levels. (G) Plasma alanine aminotransferase (ALT) levels were measured using an enzyme-linked immunosorbent assay (ELISA) kit. Data are expressed as the mean±standard error of 6 mice/group. ND, normal diet; WT, wild-type; HFD, high-fat diet; ADRP, adipose differentiation-related protein. aP<0.05 vs. WT mice with ND, bP<0.05 vs. Ppara-/- mice with ND, cP<0.05 vs. WT mice with HFD.
![]()
Fenofibrate fails to maintain peroxisomal biogenesis in Ppara-/- mice
Basal mRNA expression of PPARα target genes such as Pex11, Abcd2, Abcd3, and Acox1 were significantly decreased in Ppara-/- mice liver (Fig. 4A). Basal expression of Pex7, Pex16, Pex19, and catalase mRNA were also significantly decreased in Ppara-/- mice liver (Fig. 4A). Functional peroxisome estimated by ABCD3 and catalase immunostaining were significantly decreased in Ppara-/- mice liver (Supplementary Fig. 4). HFD feeding decreased the mRNA levels of Pex3, Pex13, Pex14, and Pex16 in Ppara-/- mice liver, which was not affected by fenofibrate (Fig. 4B).
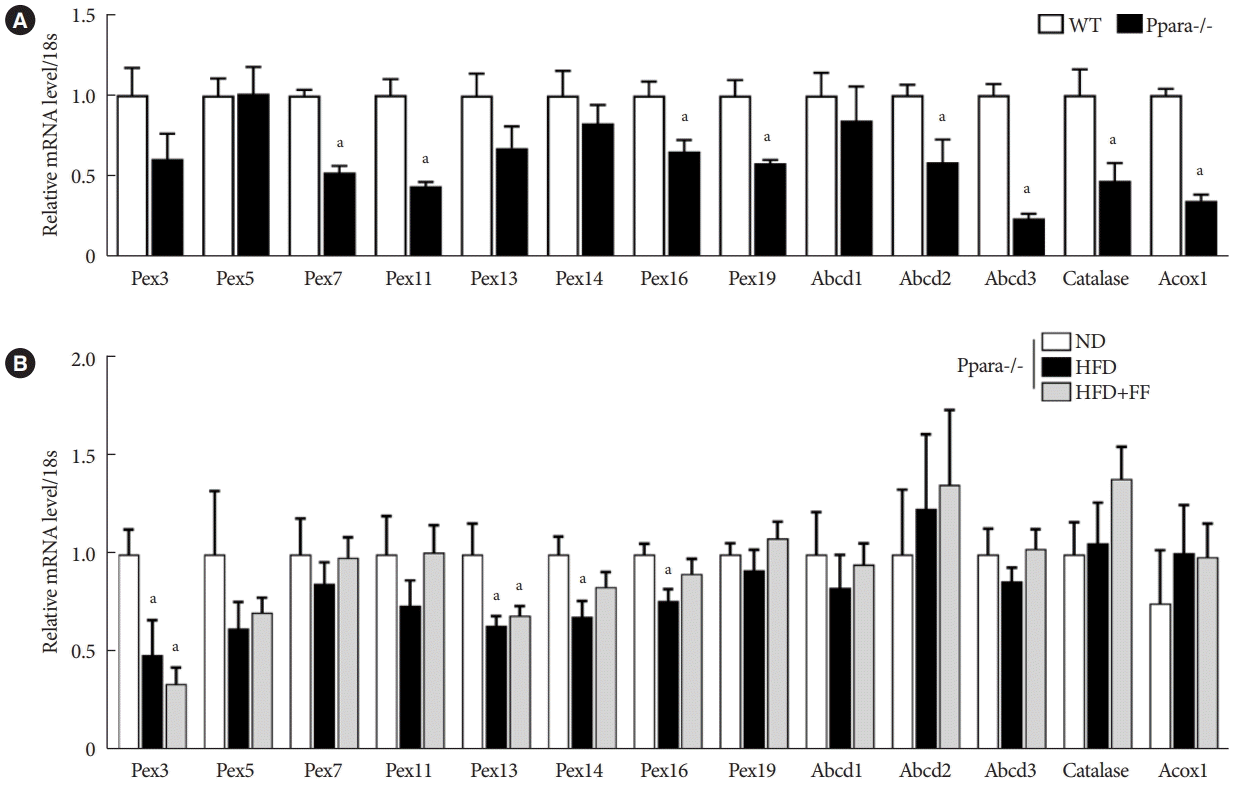
Fig. 4.
Fenofibrate fails to maintain peroxisomal biogenesis in Ppara-/- mice. (A) The expression of peroxisome-related genes was decreased in Ppara-/- mice compared to wild-type (WT) mice. (B) Peroxisome-related genes in Ppara-/- mice were analyzed by real-time polymerase chain reaction, and the results were normalized to 18S rRNA levels. Data are expressed as the mean±standard error of 6 mice/group. PPAR, peroxisome proliferator-activated receptor; Pex, peroxisomal biogenesis factor; Abcd, ATP binding cassette subfamily D member; Acox1, acyl-CoA oxidase 1; ND, normal diet; HFD, high-fat diet; FF, fenofibrate. aP<0.05 vs. WT mice or Ppara-/- mice with ND.
![]()
Fenofibrate fails to improve peroxisomal function in Ppara-/- mice
IF staining showed that ABCD3 and catalase expression were reduced in HFD-fed Ppara-/- mice compared to ND-fed Ppara-/- mice. As expected, there was little effect of fenofibrate on ABCD3 or catalase protein expression in HFD-fed Ppara-/- mice (Fig. 5A-C). Basal peroxisomal FAO in Ppara-/- mice liver was remarkably suppressed (Fig. 5D) compared to that of WT (Fig. 2G), and there was little difference in FAO among ND, HFD, and fenofibrate-treated HFD Ppara-/- mice (Fig. 5D). These data suggest that fenofibrate fails to maintain peroxisomal function in Ppara-/- mice.
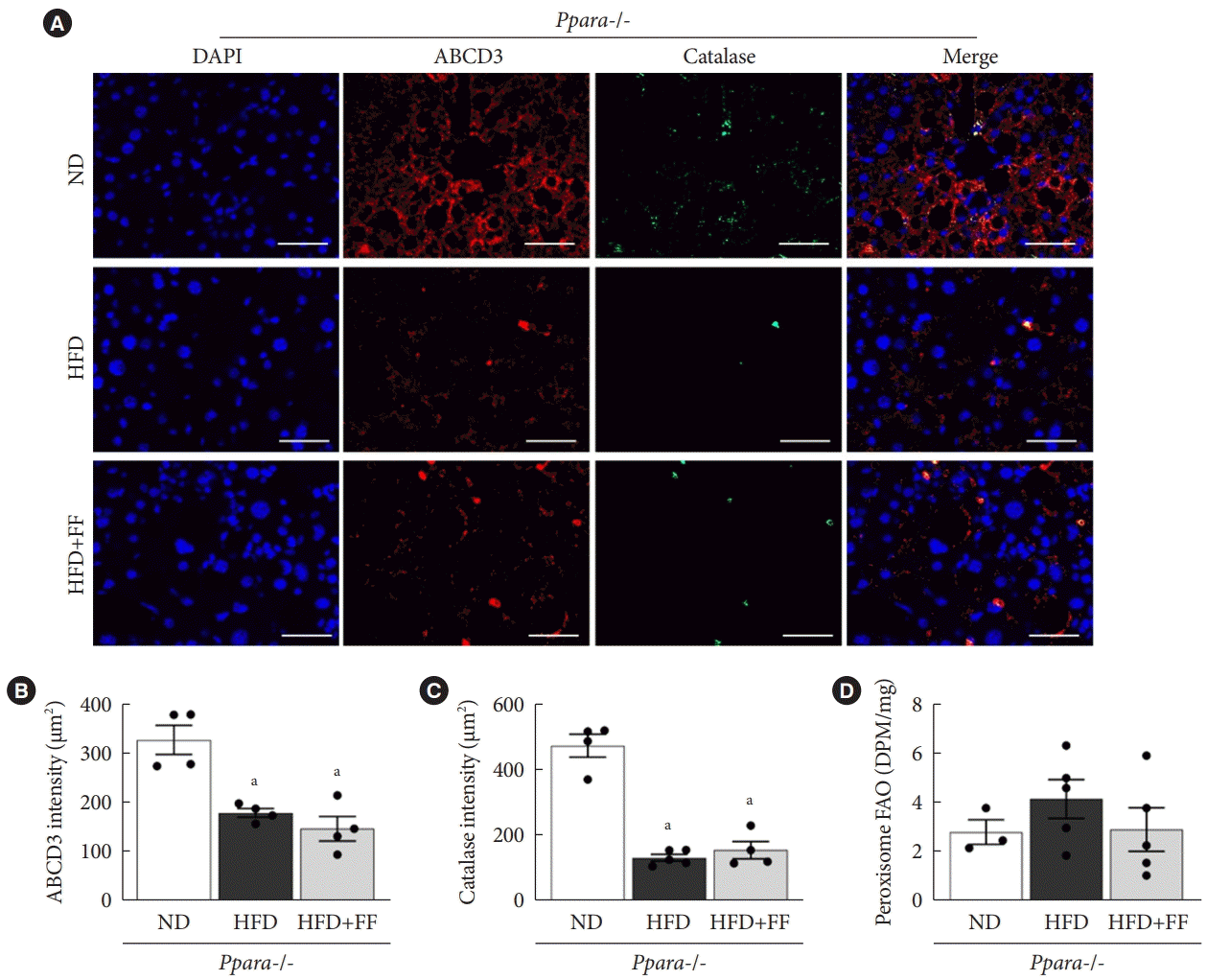
Fig. 5.
Fenofibrate fails to improve peroxisomal function in Ppara-/- mice. (A, B, C) Liver sections were immunofluorescence stained for ATP binding cassette subfamily D member 3 (ABCD3; red), catalase (green), and 4´,6-diamidino-2-phenylindole dihydrochloride (DAPI) nuclear counterstaining (blue) and intensities were quantified. Original magnification, 200×; scale bar, 200 μm. (D) Peroxisomal fatty acid oxidation (FAO) was measured in liver tissue. Data are expressed as the mean±standard error of 6 mice/group. PPAR, peroxisome proliferator-activated receptor; ND, normal diet; HFD, high-fat diet; FF, fenofibrate; DPM, disintegration per minute. aP<0.05 vs. Ppara-/- mice with ND.
![]()
DISCUSSION
In this study, administration of fenofibrate to HFD-fed mice (1) effectively prevented liver steatosis and injury characterized by ALT, inflammation, oxidative stress, and lipid accumulation; (2) significantly increased the expression of liver peroxisomal antioxidant- and biogenesis-related markers; and (3) increased liver peroxisomal FAO. In Ppara deficient mice, fenofibrate failed to maintain peroxisomal biogenesis and FAO in HFD-induced liver injury. Taken together, the present data suggest that fenofibrate improves peroxisomal fitness by increasing peroxisomal biogenesis and function and thus protects against HFD-induced NAFLD.
Extensive studies have revealed that fenofibrate decreases HFD-induced plasma TG, ALT, and insulin levels in mice [27,35,36]. In addition, fenofibrate decreases HFD-induced lipid accumulation, inflammation (Il1b, Il6, Mcp1, and tumor necrosis factor-α), oxidative stress, and fibrosis (α-smooth muscle actin [α-SMA] and COL1) in liver [27,35,36]. In the liver of HFD-fed mice, fenofibrate suppresses lipogenesis makers (sterol regulatory element-binding protein 1 [SREBP1] and ACC) [37], increases lipolysis markers (adipose triglyceride lipase [ATGL]) [38] and β-oxidant marker (ACOX1) [36]. In line with these previous reports, the present study has shown that HFD-induced liver lipid drops and plasma ALT levels were decreased in response to fenofibrate treatment. In addition, HFD-induced liver inflammation, oxidative stress, and fibrosis were reduced by fenofibrate. We also confirmed that fenofibrate increased ACOX1 expression in the liver of HFD mice, suggesting increased FAO in response to fenofibrate. However, concerns on the effect of fenofibrate on liver lipid accumulation have been reported; mice under ND treated with fenofibrate for 10 days showed increased liver TG [39], and fenofibrate simultaneous induced FAO, FAS, and FA elongation in the liver of mice under ND [40]. It remains to be important to understand the underlying mechanism of this contradictory effect of fenofibrate on liver lipid accumulation in order to develop effective strategies against NAFLD.
In this context of fenofibrate’s contradicting effect, fenofibrate increases cell viability along with upregulation of nuclear factor erythroid 2–related factor 2 (NRF2) and antioxidant enzymes only under stress condition (high glucose or hypoxia-reoxygenation injury) not at basal in cultured cardiac myocytes [41]. There are differences in activation of PPARα and the hypolipidemic effects of fenofibrate in fish between HFD and ND [42], which could be a reference for other species.
Since fenofibrate is a ligand for PPARα, we have focused on the peroxisome. More than 14 proteins are involved in peroxisomal biogenesis [11]. PEX3, PEX16, and PEX19 play important roles in the early stages of peroxisomal biogenesis, membrane integrity, and protein transport across membranes [43-45]. Also, ABCD3 has been suggested to be involved in metabolic transport of long-chain acyl-CoA [32]. Analysis of Gene Expression Omnibus, a public database, showed that PEX16 and PEX19 were significantly reduced in patients with NASH (GES164760). PEX13, a peroxisome membrane protein that helps import proteins into the peroxisome, was also significantly reduced in patients with NASH (GSE17470). In addition, deficiency of Pex11a, a peroxin involved in peroxisomal division and proliferation [46], reduces peroxisomal biogenesis and FAO, contributing to increased lipid accumulation in the liver [47]. Newborn Pex2 knockout mice exhibit cholesterol synthesis in the liver [13]. In the present study, fenofibrate increased the expression of Pex5, Pex7, Pex11, Pex13, Pex16, Pex19, Abcd2, Abcd3, and Acox1 in the liver of HFD-fed mice, suggesting that fenofibrate increases peroxisomal biogenesis and function in the liver. Since peroxisomal biogenesis is regulated by de novo biogenesis, the growth and division of pre-existing peroxisomes, and pexophagy [9], it will be interesting to understand how these three pathways govern fenofibrate-induced peroxisomal biogenesis.
The main metabolic functions of peroxisomes in mammalian cells include β-oxidation of VLCFA and ROS metabolism [12]. Catalase is the most abundant peroxisomal antioxidant enzyme [31] and effectively removes H2O2 produced during peroxisomal β-oxidation, maintaining both the cellular and the peroxisomal redox homeostasis [48]. Recent studies have demonstrated that endogenous catalase exerts beneficial effects in protecting against liver injury, including lipid accumulation and inflammation, by maintaining the liver redox balance from the early stage of HFD-induced metabolic stress [49] and NAFLD [50]. Inhibition of catalase activity augmented mitochondrial ROS production and DNA damage and impaired cell growth in human diploid fibroblasts [51]. In addition, catalase deficiency enhanced diabetic kidney injury through peroxisomal dysfunction [52]. In the present study, catalase estimated by immunostaining was increased in the liver of HFD-fed mice by fenofibrate. Although we have not measured catalase activity in the present study, decreased oxidative stress in the face of increased peroxisomal FAO in fenofibrate-treated HFD mice suggests that fenofibrate increases functional peroxisomes in the liver under HFD stress. The present data were obtained at 12 weeks after HFD feeding with or without fenofibrate; it is necessary to determine the peroxisomal biogenesis and function including FAO over time, rather than a single instant, for a comprehensive understanding.
PPARα is highly expressed in the liver and brown adipose tissue and is a key regulator of FAO [20]. PPARα is associated with peroxisomal lipid oxidation and synthesis [53]. PPARα activation not only increases the expression of FAO genes but also molecules regulating peroxisomal biogenesis in the liver [54]. When PPARα is activated, it increases the expression of Pex11, which is involved in peroxisome biosynthesis by promoting the division of peroxisomes [55]. In addition, PPARα governs inflammatory process, mainly by trans-repression of proinflammatory genes [33,36], and lipid accumulation and inflammation are intertwined [56]. Pharmacological activation of PPARα also prevents intrahepatic inflammation and fibrosis by inhibiting activated macrophages and stellate cells and lowering the expression of fibrotic markers [33]. In the liver, PPARA gene expression was significantly decreased in patients with NASH compared to those without NASH (Supplementary Fig. 2). In our study, the deficiency of Ppara augmented liver injury by increasing plasma ALT, liver inflammation and fibrosis, and lipid accumulation even under ND. In addition, the deficiency of Ppara failed to maintain peroxisomal biogenesis and function. Thus, the present data suggest a potential role of PPARα in maintaining liver homeostasis and peroxisomal fitness. However, it should be noted that there is a PPARα-independent effect of fenofibrate [23,26,27].
Further investigations are necessary to support the current findings. To confirm the involvement of the peroxisome in NAFLD, knockdown of major genes related to peroxisomal biogenesis and function need to be performed. Although delayed treatment with fenofibrate protects against HFD-induced kidney injury [23], the therapeutic effects of fenofibrate on peroxisomal biogenesis and function against NAFLD need to be investigated. Although PEX13, 16, and 19 were significantly reduced in patients with NASH, data about the involvement of peroxisomes in NAFLD in humans are not available yet.
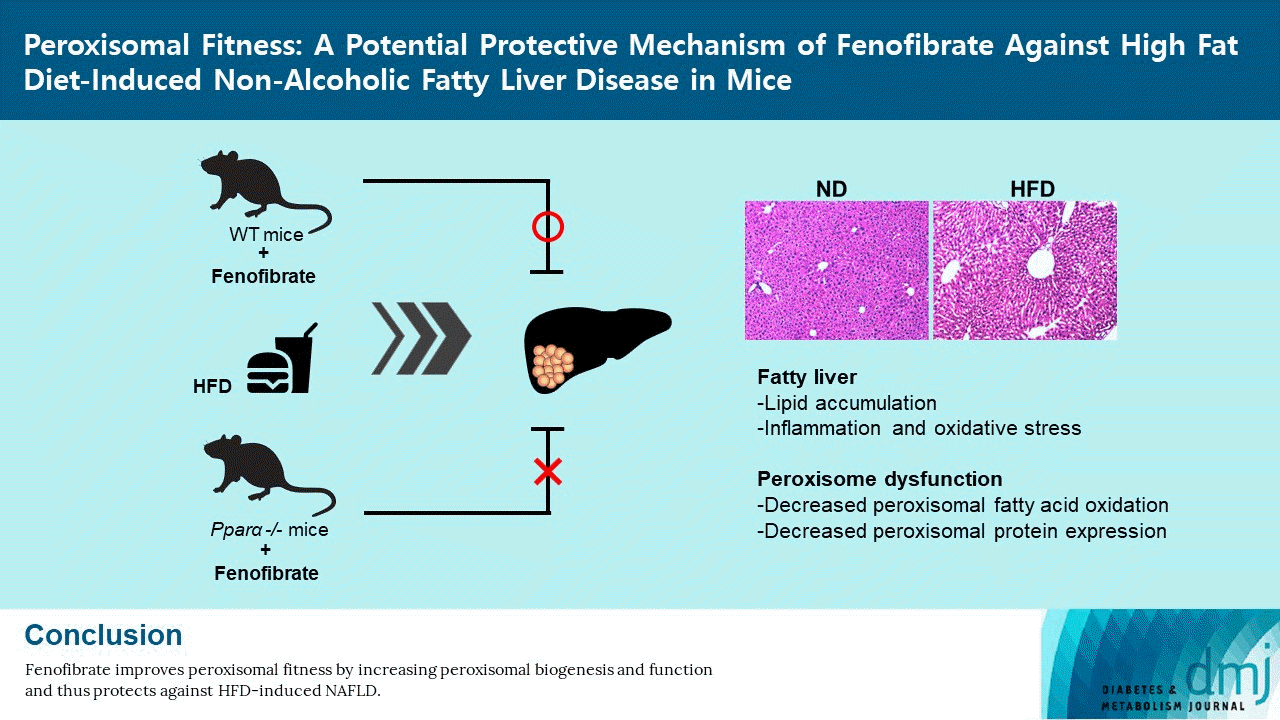
In conclusion, the present results confirm that fenofibrate protects against HFD-induced liver injury such as inflammation, oxidative stress, fibrosis, and lipid accumulation in mice. Importantly, fenofibrate increases peroxisome biogenesis and function via PPARα in the liver of HFD mice (Fig. 6). Thus, it is suggested that improved peroxisomal fitness induced by fenofibrate may play an important role in protecting against NAFLD.
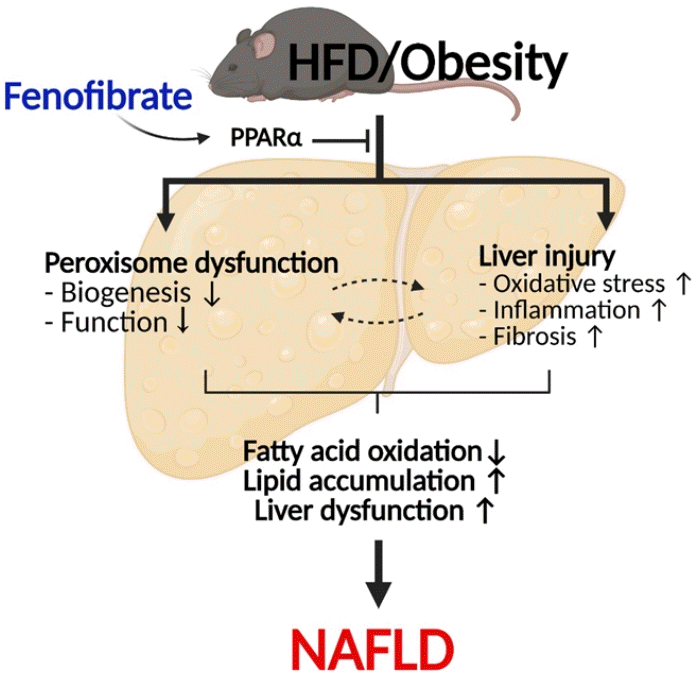
Fig. 6.
Suggested model of fenofibrate-mediated peroxisomal fitness against high-fat diet (HFD)-induced non-alcoholic fatty liver disease (NAFLD). HFD or obesity decreases peroxisomal biogenesis and function and increases liver injury, including oxidative stress, inflammation, and fibrosis, due to inhibition of the peroxisome proliferator-activated receptor α (PPARα) pathway. Subsequently, it results in decreased hepatic fatty acid oxidation, increased lipid accumulation, and the induction of liver dysfunction, which leads to the development of NAFLD. Fenofibrate maintains peroxisomal biogenesis and function through activation of the PPARα pathway. It also attenuates liver injury and increases hepatic fatty acid oxidation. Thus, fenofibrate may mediate protective effects against NAFLD by maintaining peroxisomal biogenesis and function.
![]()
 XML Download
XML Download