

 PDF
PDF Citation
Citation Print
Print



Idiopathic hypertrophic pachymeningitis (IHP) is a rare disease involving localized inflammatory thickening of the intracranial or spinal dura mater without an identified cause.1 The prevalence of hypertrophic pachymeningitis (both secondary and idiopathic) is known to be 0.949 per 100,000, with IHP reportedly constituting only half of the cases.1 The main effects of mechanical compression of nervous system structures by a thickened dura mater in patients with IHP are headache, cerebellar dysfunction, and cranial nerve palsy.2 Seizure is a very unusual presentation of IHP.2 Here we report a rare case of seizures in a patient with IHP.
CASE
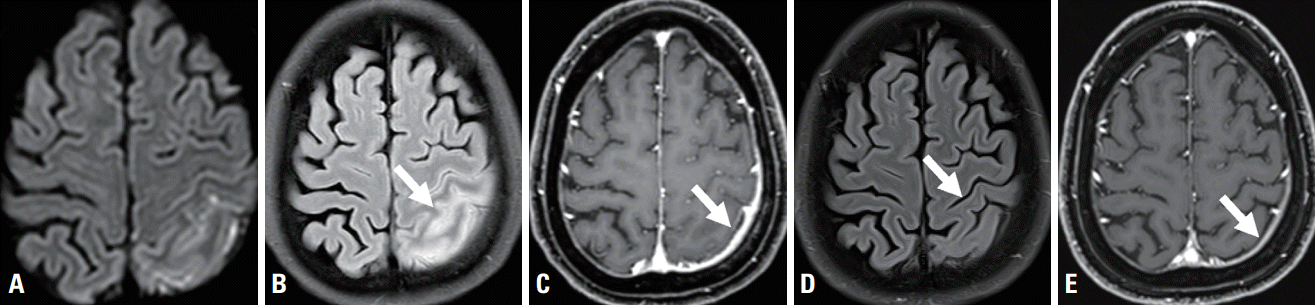
A 58-year-old female with no specific medical history visited the emergency room due to altered mentality. The emergency-room doctor reported that she presented with impaired awareness and forced right-sided eyeball deviation upon arrival at the emergency room. Deviation of the neck to the right subsequently also appeared, followed by bilateral tonic-clonic seizures. Treatment with intravenous lorazepam resulted in the seizures disappearing. Routine blood tests revealed no abnormalities other than elevation of C-reactive protein (CRP) to 1.37 mg/dL (normal range, 0.03-0.30 mg/mL). Electroencephalography performed on the day following the seizures showed a regional slow wave in the left frontocentral area. Brain magnetic resonance imaging (MRI) demonstrated pachymeningeal thickening and enhancement with gyral swelling and restricted diffusion in the left parietal lobe (Fig. 1A-C). Her cerebrospinal fluid (CSF) showed mild leukocytosis at 7/μL (normal range, 0-5/μL) and a normal protein level of 32.0 mg/dL (normal range, 15-45 mg/dL). CSF culture with antibody tests produced unremarkable findings for bacteria (Streptococcus pneumoniae, Haemophilus influenzae type B, Neisseria meningitidis, group B streptococcus, and Listeria monocytogenes), tuberculosis, fungi (cryptococcus), viruses (herpes simplex virus, varicella zoster virus, cytomegalovirus, Epstein-Barr virus, and enterovirus), and syphilis. Serum immunological tests showed values within the normal ranges for antinuclear antibodies, lupus anticoagulant, rheumatoid factor, proteins C and S, anticardiolipin antibody, anti-beta-2-GPI antibody, anti-SS-A/Ro antibody, anti-SS-B/La antibody, anti-SCL-70 antibody, anti-Jo-1 antibody, anti-Smith antibody, antineutrophil cytoplasmic antibody, complement, and IgG subclass G4. The consciousness of the patient gradually improved. She was also treated with levetiracetam at 1,500 mg per day without immunosuppressive therapy, which resulted in no more seizures. Brain MRI performed 3 months later showed improvement of the pachymeningeal thickening and enhancement with a normalized parenchymal lesion (Fig. 1D, E).
DISCUSSION
Hypertrophic pachymeningitis is a rare fibrosing inflammatory disease featuring localized or diffused thickening of the cerebral and/or spinal dura mater. It can be caused by infection, neoplasm, vascular abnormality, trauma, and systemic immunological disorders, but the cause cannot be identified in some cases, which is called IHP.3 Seizure is an uncommon manifestation of IHP.2 To the best of our knowledge, there are only two previous case reports of seizures in IHP patients,4,5 and a recent retrospective study in China found that seizures occurred in only one of 32 patients with IHP.1
IHP is considered an inflammatory or autoimmune disease, but it has not been precisely identified. An affected patient has edema in the left parietal lobe around the affected dura. This is called pachymeningoencephalitis, and is presumed to be caused by ischemia or venous congestion due to the cortex being compressed by pachymeninges, or the spreading of inflammatory cells through the subarachnoid or Virchow-Robin space. It is thought that this causes the seizures.2 A previous report indicated that perilesional edema is found in only one in 12 patients with IHP. The risk factors still need to be identified.6
When diagnosing IHP, it is necessary to identify the cause of hypertrophic pachymeningitis using blood and CSF tests, but this did not reveal the cause in our patient. Although serum CRP and CSF leukocytes were slightly elevated, this might also occur after seizures.7 The dura mater, where the blood-brain barrier is absent, may be the initial site of inflammation in autoimmune encephalitis, especially anti-N-methyl-D-aspartate receptor encephalitis.8 The probability of being diagnosed with autoimmune encephalitis may have been lower in our patient since her seizures were controlled without additional symptoms and the MRI findings improved despite immunosuppressive agents (e.g., steroids or immunoglobulins) not being administered. The gold standard for diagnosing IHP is a dura matter biopsy, which is performed when a patient shows worsening symptoms or when imaging shows aggravation despite treatment. The histopathology of IHP is characterized by the infiltration of small mature lymphocytes, plasma cells, macrophages, and epithelioid histiocytes at the surface of the dura mater. Necrotizing vasculitis of small arteries located in the dura and cerebral surfaces has also been reported.1 As in our case, a biopsy can be avoided if the symptoms improve with treatment.6
There is no consensus treatment for IHP. Steroids can be tried first, with other immunosuppressive drugs such as methotrexate or azathioprine added if there is no response to steroid therapy or if symptoms recur when the steroid dose is decreased.9
There are two recent case studies of patients with IHP who developed focal status epilepticus similar to that in our patient. All three patients showed an improvement in symptoms using steroids with or without other immunosuppressants or anticonvulsants.2,9 However, the symptoms and imaging findings of our patient improved with only anticonvulsants, and so additional immunotherapy was not considered. This case is important since it demonstrates that improvement can occur without the use of immunosuppressants. Since immunosuppressants such as steroids can have various side effects, it is necessary to carefully decide whether immunotherapy is necessary for seizure patients with IHP.
IHP is a rare cause of seizures, but the possibility of IHP should be considered in patients with the new onset of seizure symptoms. The exact mechanism underlying IHP remains unclear and guidelines for treatment have not been established, and so systematic pathophysiology-based research into IHP is necessary.
 XML Download
XML Download