

 PDF
PDF Citation
Citation Print
Print



Posterior reversible encephalopathy syndrome (PRES) and reversible cerebral vasoconstriction syndrome (RCVS) are relatively uncommon, but their diagnoses are becoming more common as brain magnetic resonance imaging (MRI) becomes both more capable and common. It has not been confirmed whether these uncommon neurological disorders can occur in the same patient. A systematic review of case series that included patients with RCVS with fatal outcomes found that about 10% of them had symmetrical high-signal-intensity lesions in the posterior regions of the brain just like in PRES.1 Although the relationship between RCVS and PRES is currently unclear, they could share a common pathophysiology. Chronic obstructive pulmonary disease (COPD) has been reported to be a rare predisposing condition for PRES development. However, RCVS has not been reported in patients with COPD. Considering that hypoxia and hypercapnia generally induce vasodilation and increase cerebral blood flow (CBF), this case report aimed to determine the pathophysiology of the co-occurrence of PRES and RCVS in a patient with an acute exacerbation of COPD (AE-COPD).
CASE
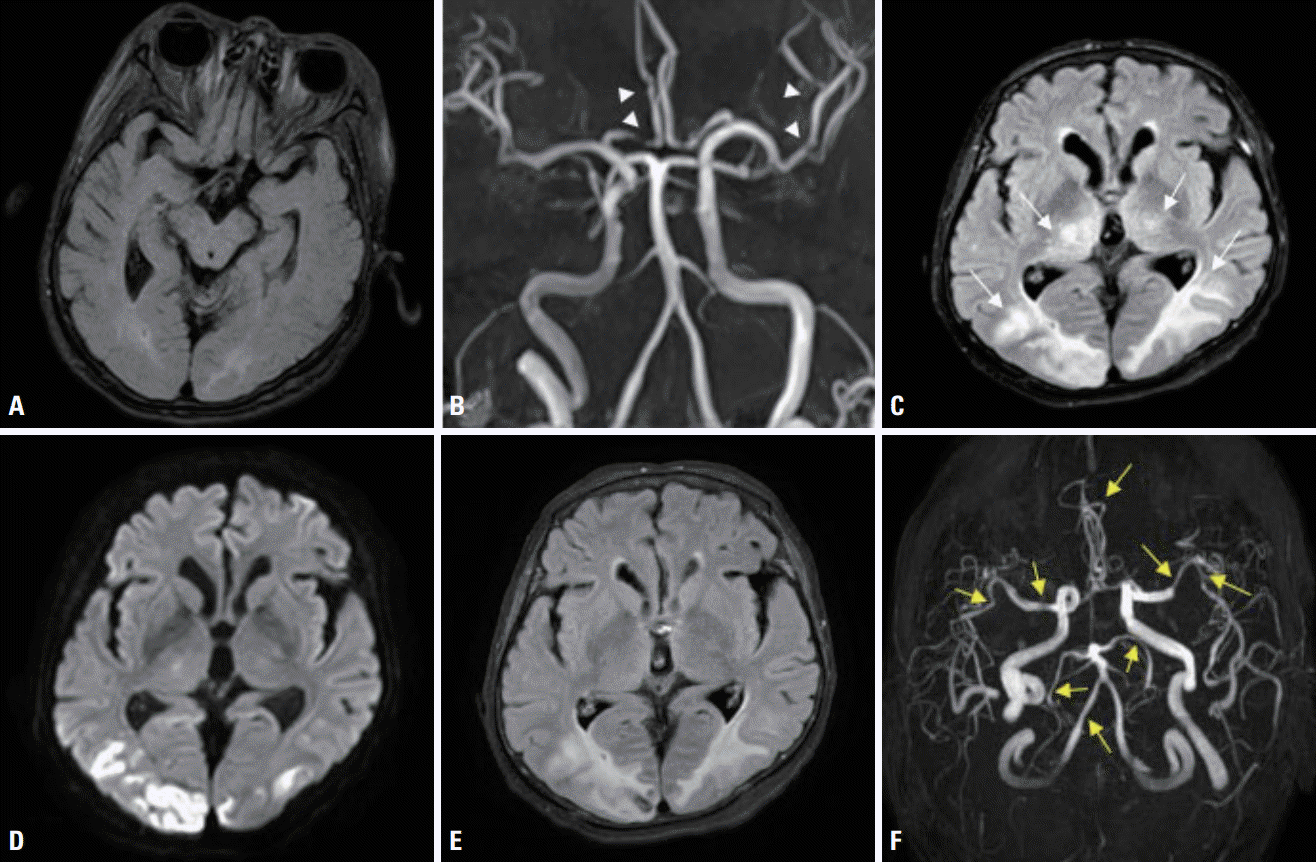
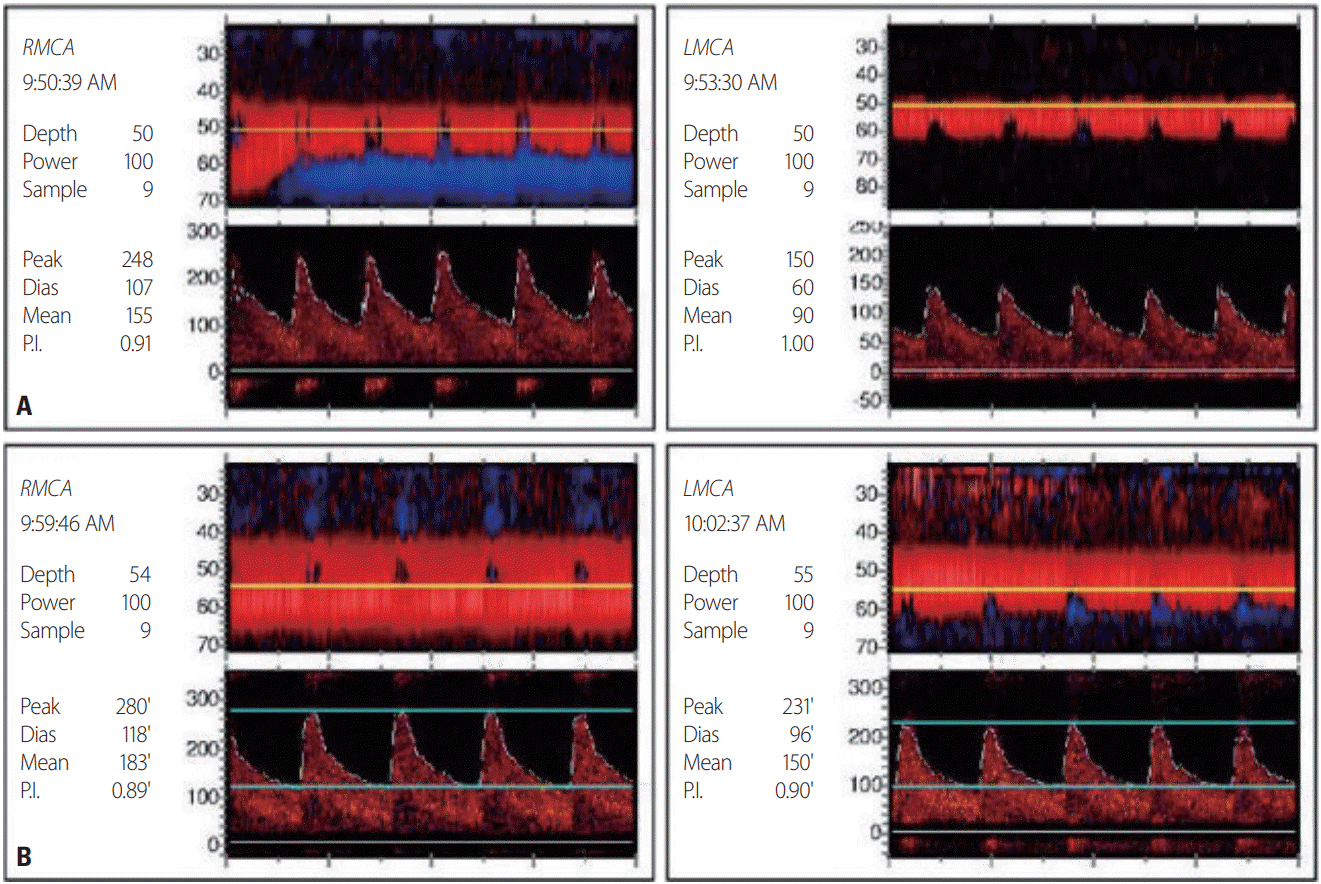
A 68-year-old female was admitted to our hospital due to seizures. The patient began experiencing a headache 11 days previously, and visited a local hospital for symptom control 2 days before the admission to our hospital. She then experienced generalized tonic-clonic seizures after clonic movement of the right upper extremity. After the seizure, she was transferred to the emergency room of our hospital. She had previously been diagnosed with COPD and had taken antitussive expectorants. Her initial blood pressure and body temperature were 141/76 mmHg and 37.0°C, respectively. A neurological examination revealed stupor, and no responses in the extremities to painful stimuli. The deep tendon reflexes were decreased slightly in both the lower and upper limbs. There was no Babinski’s sign or ankle clonus. High signal intensities were observed in both occipital lobes on an MRI fluid-attenuated inversion recovery image of her brain obtained at the local hospital (Fig. 1A). Multifocal narrowing was suspected in both the middle and anterior cerebral arteries on magnetic resonance angiography (MRA), although we did not recognize the possibility of RCVS at the time (Fig. 1B). In arterial blood gas analysis, pH was 7.20, partial pressure of carbon dioxide (PaCO2) was 106 mmHg, partial pressure of oxygen (PaO2) was 54 mmHg, and bicarbonate was 41 mmol/L. In the previous hospital records, PaCO2 was 50-60 mmHg at the time of admission, but was 120 mmHg when the seizure occurred 1 day previously. At 3 days after admission, electroencephalography (EEG) indicated frequent medium amplitude diffuse and semirhythmic delta slow waves, which peaked at the bilateral frontal areas, but without epileptiform discharge. At 10 days after admission, EEG indicated continuous diffuse and irregular delta-to-theta slow waves. At 5 days after admission, newly developed high-signal-intensity lesions with parenchymal swelling in both parietooccipital lobes and both thalami, and acute cerebral infarctions in multiple arterial territories were observed on brain MRI that was performed before restoring her consciousness (Fig. 1C-E). Clexane at 1 mg/kg was injected subcutaneously every 12 hours. There were no cardioembolic indicators on echocardiography and 24-hour Holter monitoring. The levels of D-dimer, fibrinogen, and fibrinogen degradation products were within the reference ranges. On the 1-week follow-up MRI and MRA, multifocal narrowing of intracranial arteries was observed without newly developed cerebral infarctions (Fig. 1F). Comparison with the previous MRA prompted suspicion of RCVS, and 60 mg of nimodipine was administered orally every 4 hours. The hypercapnia was aggravated at 16 days after admission. In arterial blood gas analysis, pH was 7.19, PaCO2 was 66 mmHg, PaO2 was 88 mmHg, and bicarbonate was 25 mmol/L. The mean flow velocities in both middle cerebral arteries were increased on transcranial Doppler (TCD) ultrasonography as the hypercapnia worsened (Fig. 2). After TCD ultrasonography had confirmed the worsening of vasoconstriction, nimodipine was continuously administered intravenously at 12 mL/hour. Carbon dioxide retention did not improve since her family refused the application of mechanical ventilation, nor did the vasoconstriction on TCD ultrasonography. At 39 days after admission, the patient was transferred to a local hospital, and maintained only using an oxygen mask as her family requested.
DISCUSSION
This was a very rare case in which RCVS and PRES occurred simultaneously with AE-COPD. PRES is commonly associated with high blood pressure and other antecedent diseases such as acute kidney injury, chronic kidney disease, and sepsis.2 However, the blood pressure of the patient was not very high and she did not have any of the antecedent diseases described above. Although some COPD cases with PRES have been reported without well-known antecedents, COPD cases with RCVS have not reported previously.3-6 Hypercapnia generally induces cerebral vasodilation and increases CBF, while hypocapnia induces cerebral vasoconstriction and decreases CBF. However, in the present patient with COPD, blood vessels constricted as hypercapnia worsened, as confirmed by TCD ultrasonography. COPD is an autoimmune disease that activates peripheral T-cells and increases tumor necrosis factor alpha (TNFα), interleukin-1 (IL-1), and endothelin-1 (ET-1) levels. It has been proposed that increased basal circulation of ET-1 causes vascular endothelial cell dysfunction, constricts cerebral blood vessels, and increases the permeability of the blood-brain barrier; this phenomenon may therefore lead to PRES in patients with COPD. ET-1 is further increased in acute respiratory failure, and TNFα, IL-1, and ET-1 levels are also increased in infection, which are predisposing factors for AE-COPD.3 RCVS is characterized by the variable, segmental, and multifocal vasoconstriction of the cerebral arteries. The mechanism that leads to RCVS has not been clearly identified, but the vasodilatory dysregulation of the arterial cerebral endothelium is known to play an important role. Even though most RCVS cases spontaneously resolve within 1-3 months, it has been reported that approximately 25% of patients experience complications including subarachnoid hemorrhage, convulsions, and ischemic events.7 The most common imaging findings in a recent systematic review of fatal RCVS cases were cortical subarachnoid hemorrhage, brain hemorrhage, infarction, and vasogenic edema.1 In particular, about 10% of those cases had symmetrical high-signal-intensity lesions in the posterior region of the brain just like in PRES.1 In the present case, the prospective relationship between RCVS and PRES could not be identified. However, MRI performed on the 5th and 12th days of hospitalization revealed changes in the white-matter pattern of PRES, progressive vasoconstriction, and accompanying cerebral infarction. Although the relationship between RCVS and PRES is currently unclear, we can assume that they share a common feature of cerebrovascular tone dysregulation. Endothelial cell dysfunction also inhibited normal hypoxemia and the hypercapnia-induced vasodilatory response, and was presumed to be caused by vasoconstriction.
When managing both RCVS and PRES, it is important to identify and treat their causative diseases and to interpret them with respect to autonomic dysregulation, while still controlling RCVS and PRES independently.
 XML Download
XML Download