

 PDF
PDF Citation
Citation Print
Print



INTRODUCTION
The sudden death of a young and healthy person is the most devastating scenario in clinical medicine. Coronary artery disease and acute myocardial infarction are major causes of sudden cardiac death (SCD) in older populations, and inherited cardiac disorders comprise a substantial proportion of SCD cases in patients aged 35 years and younger.1) Among inherited cardiac disorders, long QT syndrome (LQTS) is a common cause of sudden, unexplained death in young individuals.2)3) LQTS is characterized by delayed ventricular repolarization, which manifests as QT prolongation on an electrocardiogram (ECG). Lengthening of the QT interval increases the propensity for ventricular tachycardia, torsades de pointes, and sudden cardiac arrest. Over the past 20 years, significant progress has been made in its genetic and molecular biology by the identification of its causative genes.4)5) To date, molecular genetic studies have confirmed relevant mutations of 17 genes in LQTS.6) The major causative genes include KCNQ1 (LQT1), KCNH2 (LQT2), and SCN5A (LQT3), which constitute 75–80% of cases.7)8) Although the estimated prevalence is approximately 1:2500, research on Korean LQTS patients and their long-term clinical outcomes are currently sparse.9) This study aimed to characterize patients with LQTS from a clinical and genetic perspective and determine the overall outcome of patients who were registered at a single tertiary center.
METHODS
Study population
This retrospective cohort study included 105 patients diagnosed with LQTS and their genetically confirmed family members between 2000 and 2016 at the Seoul National University Hospital. The median follow-up duration was 10.5 (range, 5.3–20.0) years. Data regarding sex, age at initial symptom onset (if symptomatic), clinical presentation, age at diagnosis, current age, follow-up duration, treatment, the occurrence of cardiac events after treatment, results of genetic studies, family history, and ECG data (resting ECG, 24-hour Holter monitoring, epinephrine challenge test, or treadmill test) were collected. Schwartz scores8) were then calculated for each patient. Patients with LQTS who met the following diagnostic criteria were included: 1) proven pathogenic mutations, with or without suspicious clinical symptoms; and 2) a Schwartz score of 3.5 points or higher.
Definition
The total cohort was divided into symptomatic and asymptomatic groups, depending on the initial presenting symptoms (or lack thereof). The outcomes were evaluated and the primary outcomes were documented as overall survival and breakthrough cardiac events (BCEs). Cardiac events were defined as syncope, seizures, sustained ventricular tachyarrhythmia, or cardiac arrest. BCEs were defined as cardiac events that occurred during treatment. The targets of implantable cardioverter-defibrillator (ICD) implantation were divided into primary and secondary prevention. Among patients with LQTS who underwent ICD implantation, those with syncope refractory to pharmacological treatment without the described cardiac arrest belonged to the primary prevention group. On the other hand, ICD implantation in a patient who survived aborted cardiac arrest, documented sustained ventricular tachycardia, or ventricular fibrillation was classified as secondary prevention.
The Institutional Review Board (IRB) for Clinical Research at Seoul National University Hospital approved the use of medical records for this study (IRB H-1705-140-859) and waived the need for informed consent due to the study’s retrospective analysis of anonymized medical records.
Statistical analysis
All statistical analyses were conducted using SPSS software (version 19.0; SPSS Inc., Chicago, IL, USA). Continuous variables are expressed as means±standard deviations or median and range, whereas categorical variables are presented as frequency and proportion. Student’s t-test was used to examine continuous variables, while the χ2 test was used to analyze nominal variables. Comparisons between nonparametric parameters were performed using the Mann-Whitney U test. The survival analysis was performed using Kaplan-Meier estimation. For the subgroup analysis of survival, Mantel-Cox log-rank analysis was performed. Statistical significance was set at p<0.05.
RESULTS
Clinical characteristics
The clinical characteristics of the 105 patients with LQTS (48 women; 45.7%) are shown in Table 1. The median age at diagnosis was 11.0 (range, 0.003–80.0) years. Seventy patients were included in the symptomatic group at the initial presentation. The first clinical feature manifested at a median age of 10.5 years (range, 0.00–80.0), while the median age at diagnosis was 11.1 (0.003–80.0) years. Among them, 38 patients (54.3%) had syncope or seizures. Aborted cardiac arrest as the first presentation was observed in 30% of patients in the symptomatic group (Figure 1). Regarding phenotype and sex, 45.7% of the entire cohort were female and a significantly higher proportion of symptomatic patients were female (52.9% vs. 31.4%; p=0.038). When patients were divided into subgroups based on the age of 13 years at diagnosis, significantly more males were in the younger than 13 years group than over 13 years of age group (64.1% vs. 39.0%, respectively; p=0.012). The median corrected QT interval (QTc) was significantly longer in the symptomatic group than in the asymptomatic group (534 ms vs. 505 ms; p=0.003). The overall treatment follow-up duration was 10.5 (range, 5.3–20.0) years. One patient was initially classified as being asymptomatic but developed clinical symptoms during the follow-up period.
Table 1
Cohort characteristics and comparisons between asymptomatic patients and initially symptomatic patients
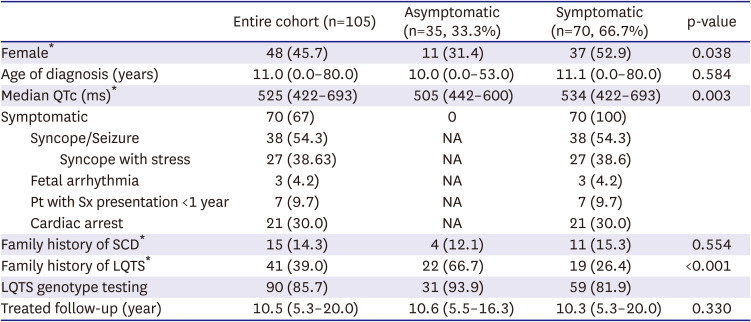
Seven patients experienced their first symptoms at 1 year of age. Three of these patients presented with arrhythmia during the fetal stage. One patient presented with neonatal ventricular tachycardia after birth and was diagnosed with LQT2. Another patient presented with fetal tachycardia and was suspected to have fetal hydrops due to ventricular tachycardia. After birth, the patient presented with torsades de pointes that had to be defibrillated. The patient was diagnosed as having LQT2 disease. Another patient presented with fetal premature ventricular beats. After birth, he presented with a prolonged QTc (619 ms), showed QT interval shortening on the lidocaine test, and was confirmed to have an SCN5A (LQT3) mutation.
Systemic syndromes associated with LQTS
Jervell Lange-Nielsen syndrome (JLNS), Andersen-Tawil syndrome, and Timothy syndrome were identified in our cohort.
Nine patients had JLNS. Their age at diagnosis was the youngest in our cohort, with a median of 4 years (range, 0.01–34.0). Among these patients, 3 presented with prolonged QTc during preoperative ECG screening for deafness-related surgery.
Of the 5 patients with confirmed Andersen-Tawil syndrome (LQT7), 3 were initially symptomatic; one showed dizziness, another complained of several syncopal events, and the other presented with dystonia. Their ECG and Holter monitoring results showed prominent U waves and frequent non-sustained polymorphic ventricular tachycardia. Systemic combined symptoms, such as hypokalemic periodic paralysis and weakness, were observed in 3 patients. All 5 patients had typical features of low-set ears, a triangular face, and micrognathia with left fifth finger clinodactyly.
In our study, 2 patients had Timothy syndrome (LQT8). They had the typical features of a dysmorphic face (round face, bald at birth, and small teeth), and syndactyly (third, fourth, and fifth digits). One patient initially presented with ventricular fibrillation under general anesthesia during syndactyly repair. ECG during sinus rhythm showed marked QT interval prolongation and a functional 2:1 atrioventricular block due to prolonged ventricular repolarization.
Genetic characteristics
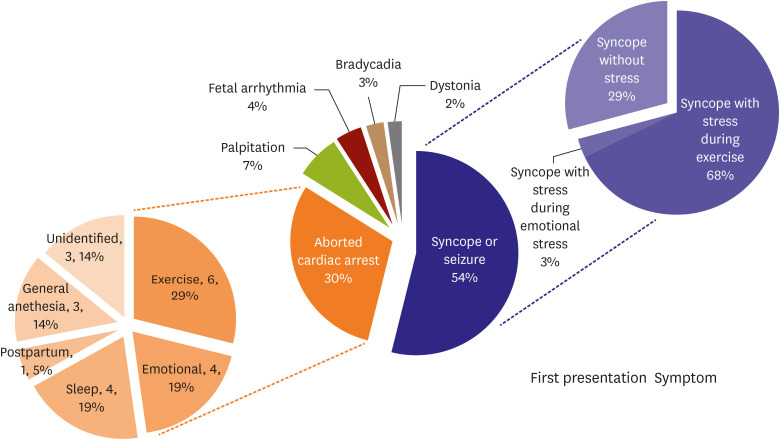
Genetic testing was performed in 90 of the 105 patients (85.7%). The 72 LQTS probands and 18 genetically proven family members were evaluated. In the family group, the penetrance of the LQTS genetic mutation was 51%. The following were tested: KCNQ1 (LQT1; n=31 [34.4%]; including JLNS [n=9, compound heterozygous mutations in KCNQ1]), KCNH2 (LQT2; n=11 [12.2%]), SCN5A (LQT3; n=11 [12.2%]), ANK2 (LQT4; n=1 [1.1%]), KCNJ2 (LQT7; n=5 [5.6%]), CACNA1C (LQT8; n=2 [2.2%]) and 3 (3.3%) of multiple LQTS-associated mutations (LQTM) (Figure 2). The genetic testing yield was 71.1%. Phenotypes according to the corresponding genotypes are listed in Table 2. There were more cases of cardiac arrest in the LQT3 group than in the LQT1 group (relative risk, 7.9, p=0.017). The proportion of patients with ICD insertion of the LQTS was significantly higher in the LQT3 group than in the other LQTS groups (p=0.001).
Figure 2
The distributions of genetic mutations are confirmed in the genetically verified group.
G−/P+ = genotype negative and phenotype positive; JLNS = Jervell Lange-Nielsen syndrome; LQT1 = congenital long QT syndrome type 1; LQT2 = congenital long QT syndrome type 2; LQT3 = congenital long QT syndrome type 3; LQT4 = congenital long QT syndrome type 4; LQT7 = congenital long QT syndrome type 7; LQT8 = congenital long QT syndrome type 8; LQTM = multiple long QT syndrome-associated mutations; VUS = variant of uncertain significance.

Table 2
Cohort characteristics and comparisons by the genotype of patients with LQTS
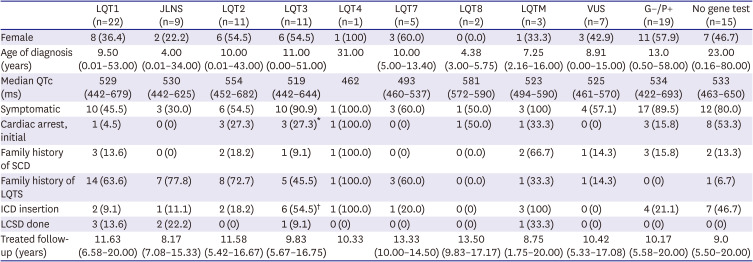
Values are presented as number (%) or median (range).
G−/P+ = genotype negative and phenotype positive; ICD = implantable cardioverter-defibrillator; JLNS = Jervell Lange-Nielsen syndrome; LCSD = left cardiac sympathetic denervation surgery; LQT1 = congenital long QT syndrome type 1; LQT2 = congenital long QT syndrome type 2; LQT3 = congenital long QT syndrome type 3; LQT4 = congenital long QT syndrome type 4; LQT7 = congenital long QT syndrome type 7; LQT8 = congenital long QT syndrome type 8; LQTM = multiple LQTS-associated mutations; LQTS = long QT syndrome; QTc = corrected QT interval; SCD = sudden cardiac death; VUS = variant of uncertain significance.
*In the LQT3 group, more people showed cardiac arrest than in the LQT1 group (relative risk, 7.9; p=0.017), by Fisher’s exact test.
†The proportion of patients with an ICD insertion of LQTS was significantly higher in those patients in LQT3 compared with all other LQTS groups (p=0.001); Bonferroni correction was applied to reduce type I error.
LQTM was detected in 3 patients. One of them was initially presented with syncope at 16 years of age and was eventually diagnosed as having KCNH2 and SCN5A mutations. Syncope usually occurred while the patient was falling asleep or resting. Another patient with medically intractable recurrent ventricular tachycardia with syncope harbored the ANK2 (LQT4) and RYR2 variants. This mutation could have caused catecholaminergic polymorphic ventricular tachycardia. Finally, the third patient had multiple associated mutations, including SCN5A and ANK2. This patient initially presented with torsades de pointes and dilated cardiomyopathy (DCM) at the age of 0.25 years.
Treatment and outcomes
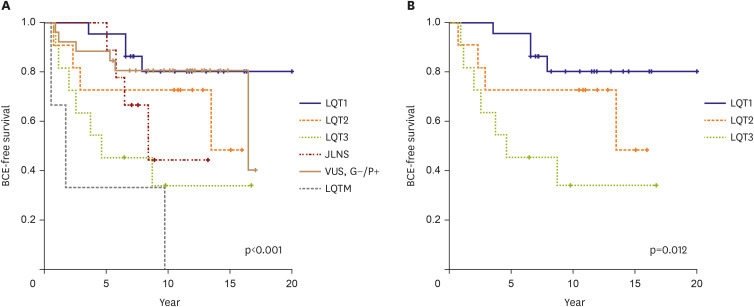
During the median 10.3 years (range, 5.3–20.0) of follow-up in the symptomatic group, β-blockers were initially administered to 60 of the 70 patients (85.7%). When BCEs occurred during follow-up, other medications were added, and if necessary, procedural or surgical treatments were considered. For the patients with SCN5A, mexiletine was added. For ANK2/RYR2, and KCNJ2 mutations associated with BCE, additional flecainide was recommended. During the follow-up, 28 patients (26.7%) in the total cohort experienced BCEs. The 10-year BCE-free survival rate was 73.2%. There was a significant difference in BCE-free survival according to the genotype (p<0.001) (Figure 3). The 1- and 10-year BCE-free survival rates were the highest in patients with LQT1, whereas the 1-year BCE-free survival rate was the lowest in patients with LQTM. When comparing LQT1, 2, and 3, which account for the majority of LQTS, the prognosis for the incidence of BCE was poor in the order of LQT3, 2, and 1 (p=0.012). In the LQT1 group, the 10-year BCE-free survival rate was 81.8%. In the entire cohort, BCEs were observed in 21 (35%) patients who received medical treatment with β-blockers alone. Two patients (0.3%) experienced breakthrough cardiac arrest during medical treatment. The risk of BCE was 3.583 times higher in women than in men (p=0.037, odds ratio, 3.583).
Figure 3
BCE-free survival curve for entire genetically verified LQTS. (A) BCE-free survival through each genotype, statistical significance was analyzed except for LQT4, 7, 8 which were rare. (B) When comparing LQT1, 2, and 3, which account for most of LQTS, the prognosis was poor in the order of LQT3, LQT2, and LQT1.
BCE = breakthrough cardiac event; G−/P+ = genotype negative and phenotype positive; LQTS = long QT syndrome; LQT1 = congenital long QT syndrome type 1; LQT2 = congenital long QT syndrome type 2; LQT3 = congenital long QT syndrome type 3; LQT4 = congenital long QT syndrome type 4; LQT7 = congenital long QT syndrome type 7; LQT8 = congenital long QT syndrome type 8; LQTM = multiple LQTS-associated mutations; VUS = variant of uncertain significance.
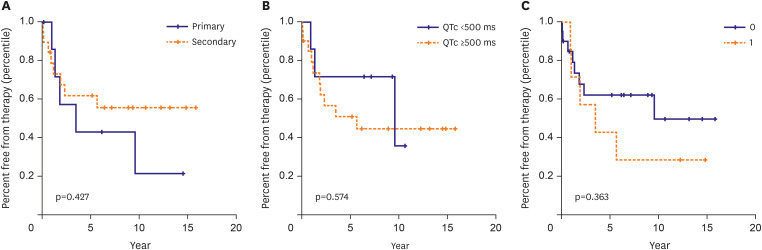
Twenty-nine patients underwent ICD implantation and/or left cardiac sympathetic denervation surgery (LCSD). ICD implantation was recommended for 28 patients, one of whom refused ICD implantation. Thus, a total of 27 patients who underwent ICD implantation were followed up for a median of 8.25 (range, 0.17–15.83) years. Of these, 17 (62.9%) were female. The median age at ICD implantation was 17 years (range; 2.0–79.0). Two patients maintained their LCSD without ICD implantation. Three patients underwent epicardial ICD implantation at the ages of 2.3, 6.0, and 7.8 years, and all 3 weighed less than 20 kg. Our study showed that LQT3 patients had a higher probability of undergoing ICD implantation than patients with other genotypes (p=0.001). Among the 6 patients with LQT3 who underwent ICD implantation, 2 received dual-chamber ICD. Another mixed group of patients with SCN5A and ANK2 mutations received a dual-chamber ICD due to sinus bradycardia. Eight patients (29.6%) underwent ICD implantation for primary prevention, whereas 19 (70.4%) underwent ICD implantation for secondary prevention. Among the primary prevention recipients, one with LQT1 underwent LCSD before ICD. Thirteen patients (48.1%) with ICDs received the appropriate shock therapy. The Kaplan-Meier analysis for appropriate shock therapy was significantly different according to the LQTS type (p=0.001). Patients with LQT2 were most likely to receive the appropriate shock therapy. Primary versus secondary prevention, QTc >500 ms, and a family history of SCD were not significantly associated with appropriate shock therapy (Figure 4).
Figure 4
Individual/cumulative risk factors for appropriate ICD shock. Among the ICD recipients, the Kaplan-Meier curves were analyzed through the risk factors, primary vs. secondary prevention (A), QTc ≥500 ms (B), and family history of sudden cardiac death (C) for the event that received appropriate shock treatment.
ICD = implantable cardioverter-defibrillator; QTc = corrected QT interval.
LCSD was performed in 7 patients (10% of the 70 patients in the symptomatic group). The median age at LCSD was 13.7 (range, 2.6–34.4) years. The median follow-up duration was 13.42 (range, 7.5–20.0) years. All the LCSD procedures were performed using video-assisted thoracoscopy. There were no LCSD-related complications, especially Horner’s syndrome. There were 5 patients with LQT1 (including 2 with JLNS), one patient with LQT3, and one patient with LQTM (LQT4 with concomitant RyR2 mutation). Among the 7 LCSD patients, 2 (1 with LQT1, 1 with JLNS) performed well with LCSD only under medical therapy, and 3 (2 with LQT1, 1 with JLNS) required subsequent ICD due to further BCEs. The other 2 patients (1 with LQT3 and the other with LQTM) underwent LCSD after ICD implantation. Up to date, there have been no further BCEs.
All except one of our patients were alive, and the patient who died of heart failure was among the aforementioned LQTM (SCN5A and ANK2 mutation) patients. The patient underwent ICD implantation at the age of 27 months. He had a family history of sudden death, and his older brother died at a young age. Although he received 2 appropriate shocks for ventricular fibrillation, he eventually died of DCM. One LQT3 patient underwent cardiac transplantation for progressive DCM.
DISCUSSION
Our cohort of single-center Korean patients with LQTS had good outcomes, except for one death. The 10-year BCE-free survival rate was 73.2% in the entire cohort and 81.8% in the LQT1 cohort. Previous studies have shown that advances in the understanding of pathogenesis, diagnostic methods, and optimal treatments have resulted in better outcomes.10)11)12)13)14) Robust genetic diagnosis, risk strategy, and proper treatment are recommended for a better prognosis.
The proportion of female patients was significantly higher in the symptomatic group (52.9%) than in the asymptomatic group (31.4%). Although LQTS is an autosomal dominant or recessive inherited trait, there are sex-based differences in its manifestation. In this study, 64.1% of the patients under 13 years of age were male, whereas 61% of the patients over 13 years of age were female. Several studies have demonstrated similar results, and this risk is associated with sex-based differences, which might be due to hormonal effects.15)16)17)18)19)20) This suggests that different approaches and treatment strategies are required according to age and genotype.
The prognosis was poor among patients with LQT3 and LQTM. This trend was similar to that observed in a study of 166 patients with LQTS published by the Mayo Clinic in 2017.15) Moreover, in our cohort, one LQT3 patient and one LQTM (SCN5A and ANK2 mutations) with TAZ mutation patient showed progressive left ventricular dysfunction and DCM. The pathogenic variant of TAZ is the causative substrate of infantile cardiomyopathy.21)22) Multiple LQTS mutations as well as concomitant mutations in cardiomyopathy may lead to a poorer prognosis. Lifestyle modification and management strategies informed by genetic evaluation may effectively improve treatment outcomes.
Guidance on lifestyle modification is important for the management of patients with LQT phenotypic characteristics. In addition, as drug treatment, several β-blocker priorities have been proposed based on the genotype-phenotype correlation.23) LQTS is an ion channelopathy, and depending on the associated pathogenesis, for example, mexiletine effectively treats LQT3.24) As a more invasive procedure, LCSD is also suitable for treating LQT1.25)26) At our institution, as a first-line drug, β-blockers were used in all groups, and in the case of LQT3, the addition of mexiletine was recommended. For LQT2 and LQT7 patients, the potassium level should be normal and supplementation may be required. Despite pharmacological management, if there are symptoms of syncope in LQT1 patients, we consider LCSD before ICD implantation, especially in children. In addition, in the case of LQT2 and LQT3, ICD was considered if breakthrough syncope occurred despite medications. These strategies are based on a genetically defined subgroup, susceptibility to sympathetic surges, and a relatively high rate of complications associated with childhood ICD.
Among the study cohort that underwent ICD implantation, nearly 29.6% received devices for primary prevention. In contrast, Horner et al.27) reported that nearly 85% of 51 patients underwent ICD implantation for primary prevention. Differences may be found in the demographic data collected from the study cohorts (our study investigated younger patients) or medico-social differences. During the median follow-up duration of 8.25 years, approximately half (48.1%) of the recipients received appropriate shock therapy. In 2 studies, patients with LQTS who received an ICD were given appropriate life-saving shock therapy while considering their risk factors, such as secondary prevention, genotype, and corrected QT.27)28) In our study, patients with LQT2 benefited from proper ICD treatment. These observations are consistent with those of the aforementioned study27) in which non-LQT3 was one of the most predictive factors for appropriate shock. However, considering the concerns of ICD implantation, better evaluation of patient-specific treatment options improves clinical outcomes.29)
LCSD is also an optimized treatment for pharmacologic non-responders.12) In our cohort, 7 patients underwent LCSD, and in some cases, medical treatment was discontinued thereafter. Nonetheless, further research is needed to determine whether LCSD alone can safely replace medical therapies.
Our study has some limitations. The most important limitation is the relatively small sample size. Due to the heterogeneous nature of genetic mutations among patients with LQTS, the sample size was insufficient for comparison with each clinical or genetic subgroup. Our study employed a retrospective cohort study design, and there were some ineligible cases in which the items for data analysis were omitted from the medical records, resulting in incomplete data. This study could also have had a selection bias, as it was based on data from a single tertiary referral center.
In conclusion, with an increased understanding of genotypes in Korean patients with LQTS, recognition of molecular pathogenesis and genetically tailored therapies has been fostered. In this study, devastating cardiac events were prevented and good long-term outcomes were achieved in patients with genetically confirmed and properly managed LQTS. Finally, further nationwide studies are needed to validate our findings.
 XML Download
XML Download