

 PDF
PDF Citation
Citation Print
Print



Introduction
Flame retardants are chemical additives in consumer products and construction materials that reduce the flammability of materials to meet fire safety standards. Until the 2000s, polybrominated diphenyl ether was one of the most common flame retardants used in plastics, foams, electronics and textiles. However, it has been found to be harmful to human health; therefore, its use was discontinued. Consequently, alternative materials such as organophosphorus flame retardants (OPFRs) have been used [1]; however, OPFRs are used as additives rather than as chemical bonds, and can easily leach into the surrounding environment [2].
Triphenyl phosphate (TPHP) is an OPFR that is widely used as a plasticizer and flame-retardant medium. The chemical structure for TPHP is (C6H5O) 3P=O, and its half-life is 1.2–5.5 years under neutral conditions [2]. Humans are commonly exposed to TPHP through hand-to-mouth contact, skin absorption, ambient air intake, and food and drinking water consumption [3]. TPHP is frequently detected in indoor dust samples from homes, offices, and vehicle interiors at relatively high levels of up to 1.8 ng/kg bw/day [4]. Foods, such as fish, and various aquatic environments, including surface and drinking water sources, are often contaminated with a relatively high concentration of TPHP [5]. A high concentration of TPHP and its primary metabolite, diphenyl phosphate (DPHP), has been detected in human tissues, including human breast milk, of which >60% of samples tested in Japan, the Philippines, and Vietnam were positive [6]. DPHP has been detected in the maternal decidua and placental chorionic villi [3], the urine of infants in >93% of North Carolina, and the urine of men and women at an average concentration of 1.3 μg/L [7]. Additionally, nail polish may be a significant source of short-term TPHP exposure and a source of chronic exposure for frequent users [8]. The 2013–2014 National Health and Nutrition Examination Survey revealed a higher concentration of TPHP in urine samples in women than in men [9].
However, little is known regarding the potential health effects of TPHP. In animals, TPHP exposure can lead to metabolic and endocrine disruption. In fish, TPHP disrupts hepatic carbohydrate and lipid metabolism [10] and alters the hypothalamic-pituitary-thyroid axis gene expression [11], which is important for growth and development. In male rodents, neonatal exposure to TPHP and DPHP for 1–10 days at 2 μg per day via subcutaneous injection showed altered lipolysis pyruvate metabolism, and the tricarboxylic acid cycle by disrupting both endocrine and noradrenergic mechanisms [12]. TPHP (40–500 μg/L) may also interfere with thyroid hormone homeostasis in rat pituitary and thyroid follicular cell lines [13].
In terms of reproductive outcomes, Japanese medaka exposed to TPHP at 0.131, 0.363, and 1.773 μg/L for 100 days showed delayed ovarian development and reduced egg production [14]. Zebrafish exposed to TPHP at 40, 200, or 1,000 μg/L for 14 days showed a significant increase in the plasma levels of 17-estradiol and testosterone [15].
Very few studies have investigated the effects of TPHP on female reproduction in humans. Carignan et al. [16] have reported that the concentrations of some urinary organophosphorus flame retardant metabolites (DEHP, bis [1,3-dichloro-2-propyl] phosphate, isopropylphenyl phenyl phosphate, tertbutylphenyl phenyl phosphate, and bis [1-chloro-2-propyl] phosphate) were negatively associated with the number of successful fertilization, implantation, clinical pregnancy, and live birth [16]. Additionally, among subfertile women, urinary DPHP metabolite concentrations measured during the assisted reproductive technology cycle of conception may be associated with early pregnancy loss [17].
Human Ishikawa endometrial cancer cells have high expression of estrogen and progesterone receptors and low histological differentiation, and are regulated similarly to that of normal endometrium. The human Ishikawa endometrial cancer cell line is a standard model for studying hormonally regulated events in the human endometrial epithelium [18]. Therefore, we used the human Ishikawa endometrial cancer cell line as a model to investigate the effects of TPHP exposure on the human endometrium. This study aimed to examine how TPHP exposure disrupts the cellular physiology of the human endometrium, as this exposure may have a detrimental effect on female reproduction.
Materials and methods
1. Culture of human Ishikawa endometrial cancer cell line
Human Ishikawa endometrial cancer cells were a kind gift from Dr. YH Kim at the Asan Medical Center in Seoul, Korea. The cells were maintained in DMEM (Gibco, Carlsbad, CA, USA) supplemented with 10% fetal bovine serum (FBS) and 1×antibiotic-antimycotic solution (penicillin 100 units/mL, streptomycin 100 μg/mL, amphotericin B 0.25 μg/mL), at 37°C under 5% CO2. Cells seeded in 24-well plates entered logarithmic growth phase after 48–72 hours. When the cells grew confluent, they were trypsinized and seeded in fresh medium. TPHP was purchased from Sigma-Aldrich (St. Louis, MO, USA). Stock solutions of TPHP were prepared with dimethyl sulfoxide (DMSO) at various concentrations that allowed an equal volume to be added to culture wells for each treatment group, to control for solvent concentration over the 48-hour incubation period. The final concentrations in the culture were 0, 10, 100, and 1,000 pmol of TPHP, which were equal to approximately 0, 0.00326, 0.0326, and 0.326 μg/L, respectively. The final concentration of DMSO in the treatment and control cultures was 0.1%.
2. MTT assay to measure TPHP dosing on human Ishikawa endometrial cancer cells
To avoid the influence of cytotoxicity on human Ishikawa endometrial cancer cells, cell viability was assessed using the MTT assay. Cells were seeded into 96-well plates and incubated for 1–3 days before the MTT assay was used to detect the cellular reduction of the water-soluble tetrazolium salt 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazoliumbromide to insoluble formazan. The cell-free supernatant was discarded, and the formazan crystals were dissolved in DMSO. Absorbance was detected at 495 nm using an enzyme linked immunosorbent assay microplate reader (BioTek Instruments Inc., Santa Clara, CA, USA). The fold-number change in living cells was determined by comparing the results with those obtained for the untreated controls. Each treatment comprised three replicates.
Unlike natural hormones, the dose response curve of an endocrine disrupting chemical (EDC) in the human body has an unpredictable characteristic, including a U or S shape, which limits the accurate determination of a specific dose [19,20]. Additionally, EDCs have been reported to have a significant effect in in vitro studies, even at low and nonmonotonic doses [19,21]. Therefore, in the present study, the dose of TPHP was determined by measuring the metabolic activity of the cells using the MTT assay.
3. RNA isolation and reverse transcriptionquantitative polymerase chain reaction (RT-qPCR)
RNA was isolated from human Ishikawa endometrial cancer cells that were seeded into 12-well plates using the Tri-RNA reagent (Favorgen, PingTung, Taiwan) following the manufacturer’s protocol. The concentration of total RNA was calculated spectrophotometrically (Bio Tek Instruments, Inc., Santa Clara), and complementary DNA was generated from the total RNA using Maxine RT PreMix (Intron, Seongnam, Korea). Quantitative PCR was performed using a 2×Brilliant III SYBR Green QPCR kit (Agilent, Santa Clara, CA, USA) on an Applied Biosystems 7000 real-time PCR system (Life Technologies, Waltham, MA, USA). All primers were purchased from BIONICS (Seoul, Korea), and are listed in Table 1. The data were analyzed after normalization to glyceraldehyde 3-phosphate dehydrogenase mRNA levels using the equation 2ΔΔCT, where ct is the cycle threshold. Quantification of each selected gene was performed using three replicate samples.
4. Protein extraction and western blotting
For total protein, cells were harvested from 6-well plates in the presence of phosphatase inhibitor cocktail III (Calbiochem Co., San Diego, CA, USA), which was added to the lysis buffer (PRO-PREP, Intron). Lysates were sonicated and centrifuged at 12,000×g for 5 minutes at 4°C. Protein concentrations were determined using Bio-Rad Protein Assay Dye Reagent Concentrate (Bio-Rad, Hercules, CA, USA).
For nuclear proteins, cells were lysed in 100 μL of hypotonic buffer (10 mM HEPES/KOH, 2 mM MgCl2, 0.1 mM EDTA, 10 mM KCl, and 1×protease inhibitor, pH 7.9), left on ice for 10 minutes, vortexed, and centrifuged at 12,000×g for 5 minutes. Pellets were washed with phosphate-buffered saline and suspended in 50 μL of ice-cold saline buffer (50 mM HEPES/KOH, 50 mM KCl, 300 mM NaCl, 0.1 mM EDTA, 10% glycerol, and 1×protease inhibitor, pH 7.9), left on ice for 2 hours, vortexed, sonicated, and centrifuged at 15,000×g for 5 minutes at 4°C.
Protein lysates were separated by sodium dodecyl sulfatepolyacrylamide gel electrophoresis and transferred onto nitrocellulose membranes. The membranes were then incubated with the corresponding primary antibodies and subsequently with their respective secondary antibodies (goat anti-rabbit or goat anti-mouse).
Among the primary antibodies, rabbit polyclonal estrogen receptor (ER) α, ER β, nuclear factor kappa-light-chain-enhancer of activated B cells (NF-κB), inhibitor of κB (I-κB), mouse polyclonal phosphoinositide 3-kinase (PI3K), peroxisome proliferator-activating receptor-γ (PPAR-γ), and β-actin were provided by Santa Cruz (Dallas, TA, USA); cyclin D1, cyclin D3, cyclin-dependent kinase (CDK) 2, p-protein kinase B (AKT), and p-p38 were provided by Cell Signaling Technology (Danvers, MA, USA). Primary antibodies were diluted at 1:1,000, and secondary antibodies were diluted at 1:2,000.
Signal detection was performed using enhanced chemiluminescence peroxidase substrate solution (ELPIs Biotechnology, Daejeon, Korea). For quantification, western blot bands were captured using an imaging system and normalized to the intensity of the corresponding β-actin band densities for statistical analysis. Each treatment comprised three replicates.
5. Immunofluorescence
Cells were grown to 80% confluence on cover slips in 6-well plates, fixed with 2% paraformaldehyde, permeabilized with 0.01% Triton X-100, both dissolved in PBS, and blocked in 10% FBS for 1 hours at room temperature. Primary antibodies (mouse polyclonal ERα and PPAR-γ and rabbit polyclonal NF-κB) were added and incubated overnight at 4°C, washed three times in phosphate buffered saline with tween 20, and incubated for 45 minutes at room temperature with secondary antibody diluted in blocking buffer. Following staining with 4′-6′diamidino-2-phenylindole dihydrochloride (DAPI), with the cells were mounted in anti-fade mounting solution (Invitrogen, Carlsbad, CA, USA) and imaged using an LSM800 confocal microscope system (Zeiss, Germany).
6. Propidium iodide (PI) staining
PI is a fluorescent intercalating agent that can be used to stain cells and nucleic acids and is used as a DNA stain in flow cytometry to evaluate cell viability or DNA content in cell cycle analysis. Cells were fixed with 70% ethanol overnight at 4°C to estimate the number of cells in each phase of the cell cycle. The fixed cells were resuspended in PI solution (50 μg/mL, 0.1 mg/mL RNase A, and 0.05% Triton X-100) and incubated for 30 minutes at 37°C, before being analyzed by flow cytometry (Zeiss, Oberkochen, Germany).
7. Statistical analysis
Statistical analyses were performed using Windows Graph Pad Prism 5 (GraphPad Software, Inc., San Diego, CA, USA). Data are expressed as mean±standard error of mean. Significance was assessed using an independent two-tailed Student’s t-test and analysis of variance. Statistical significance was set at P<0.05, indicates non-specific.
Results
1. Viability of TPHP-treated human Ishikawa endometrial cancer cells
The viability of human Ishikawa endometrial cancer cells was determined using an MTT assay. We examined the effects of increasing the exposure concentration of TPHP (0, 10, 100, and 1,000 pmol) for 24, 48, and 72 hours on cell viability. At all treatment levels tested in this study, the cells were viable. None of the tested TPHP concentrations were toxic to the cultured human Ishikawa endometrial cancer cells. Based on these results, we performed further investigations on cells treated with 0, 10, 100, and 1,000 pmol TPHP for 48 hours.
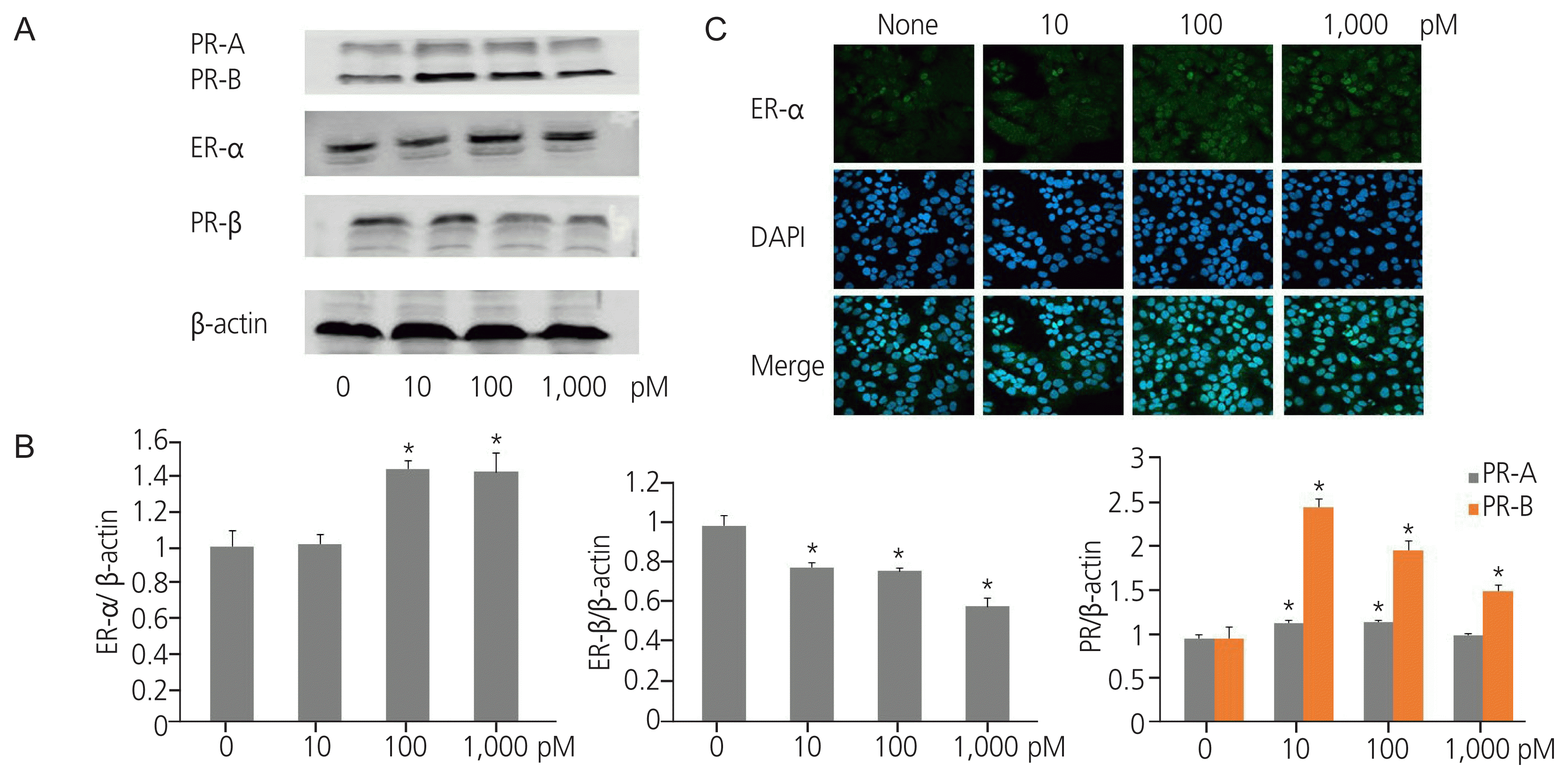
2. TPHP exposure changed ER and progesterone receptor expression in human Ishikawa endometrial cancer cells
Western blot analysis revealed that TPHP exposure elevated ER α protein levels in human Ishikawa endometrial cancer cells compared to that in vehicle-treated cells (Fig. 1A, B). Immunofluorescence analysis using ER α antibody showed a similar effect (Fig. 1C). In contrast to ER α expression, TPHP exposure reduced ER β protein levels in human Ishikawa endometrial cancer cells compared to that in vehicle-treated cells (Fig. 1B). Regarding PR expression, TPHP exposure significantly elevated PR-B, but not PR-A, protein levels in human Ishikawa endometrial cancer cells compared to that in vehicle-treated cells (Fig. 1B).
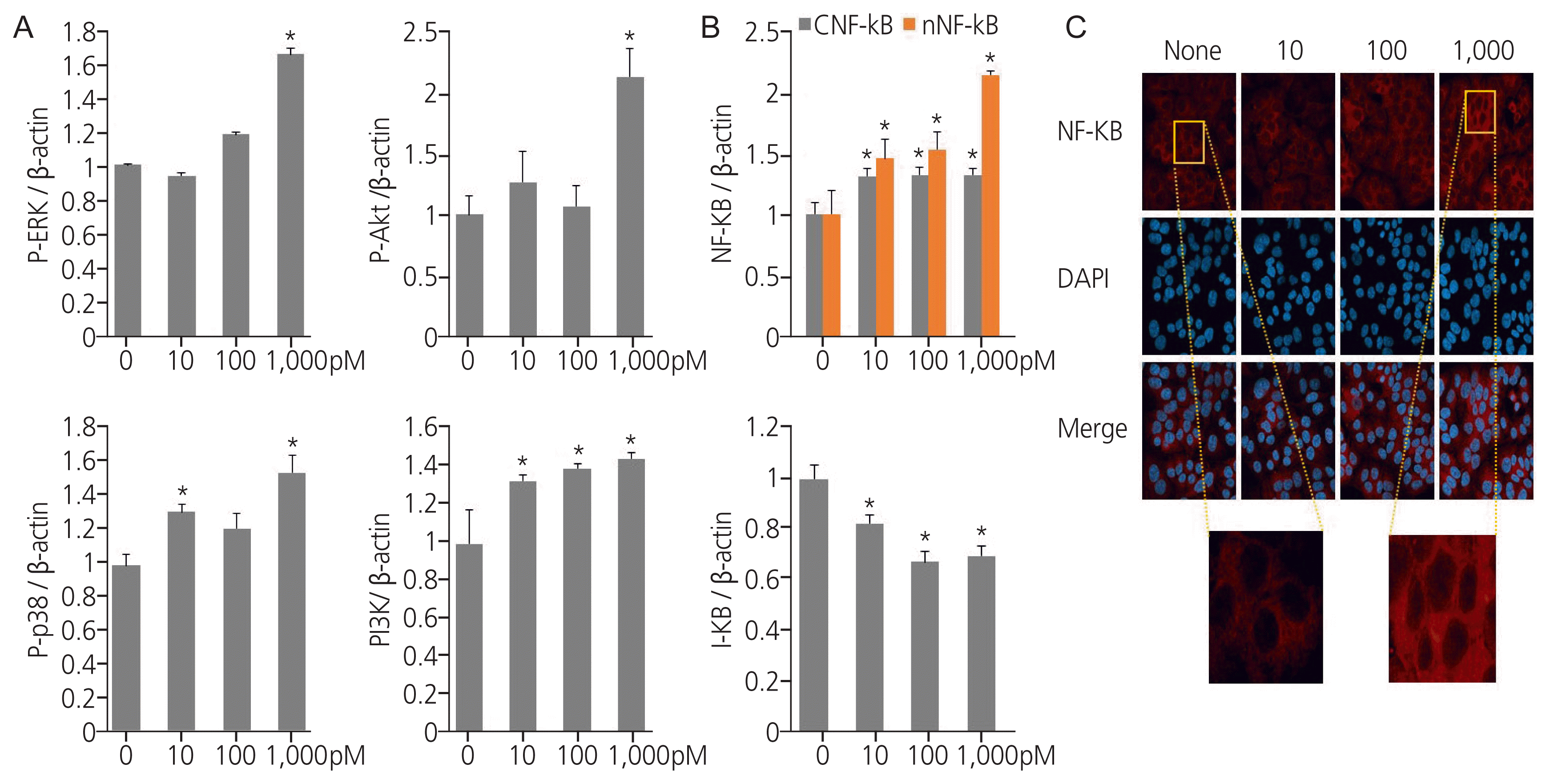
3. TPHP exposure enhances the proliferation and survival signaling in human Ishikawa endometrial cancer cells
We investigated the effect of TPHP exposure on proliferation and survival signaling in human Ishikawa endometrial cancer cells. TPHP exposure elevated the levels of p-ERK, p-AKT, and p-p38 proteins in human Ishikawa endometrial cancer cells compared to vehicle-treated cells (Fig. 2A), and a similar effect was observed for PI3K protein levels (Fig. 2A). Therefore, TPHP exposure activates the PI3K/AKT and ERK signalling pathways. Moreover, TPHP exposure enhanced the p38 signalling pathway, thereby improving survival.
4. Exposure to TPHP stimulates NF-kB signalling in human endometrial cancer cells
Exposure to endocrine disruptors modulates inflammatory pathways via the NF-κB signalling axis. To investigate the mechanism by which TPHP exerts its effects, NF-κB expression was evaluated using western blotting. Exposure to TPHP significantly increased the amount of nuclear NF-κB protein in human Ishikawa endometrial cancer cells compared to that in vehicle-treated cells, and I-κB showed degradation (Fig. 2B). Fig. 2C shows the immunofluorescence staining of NF-κB after TPHP exposure (Fig. 2C).
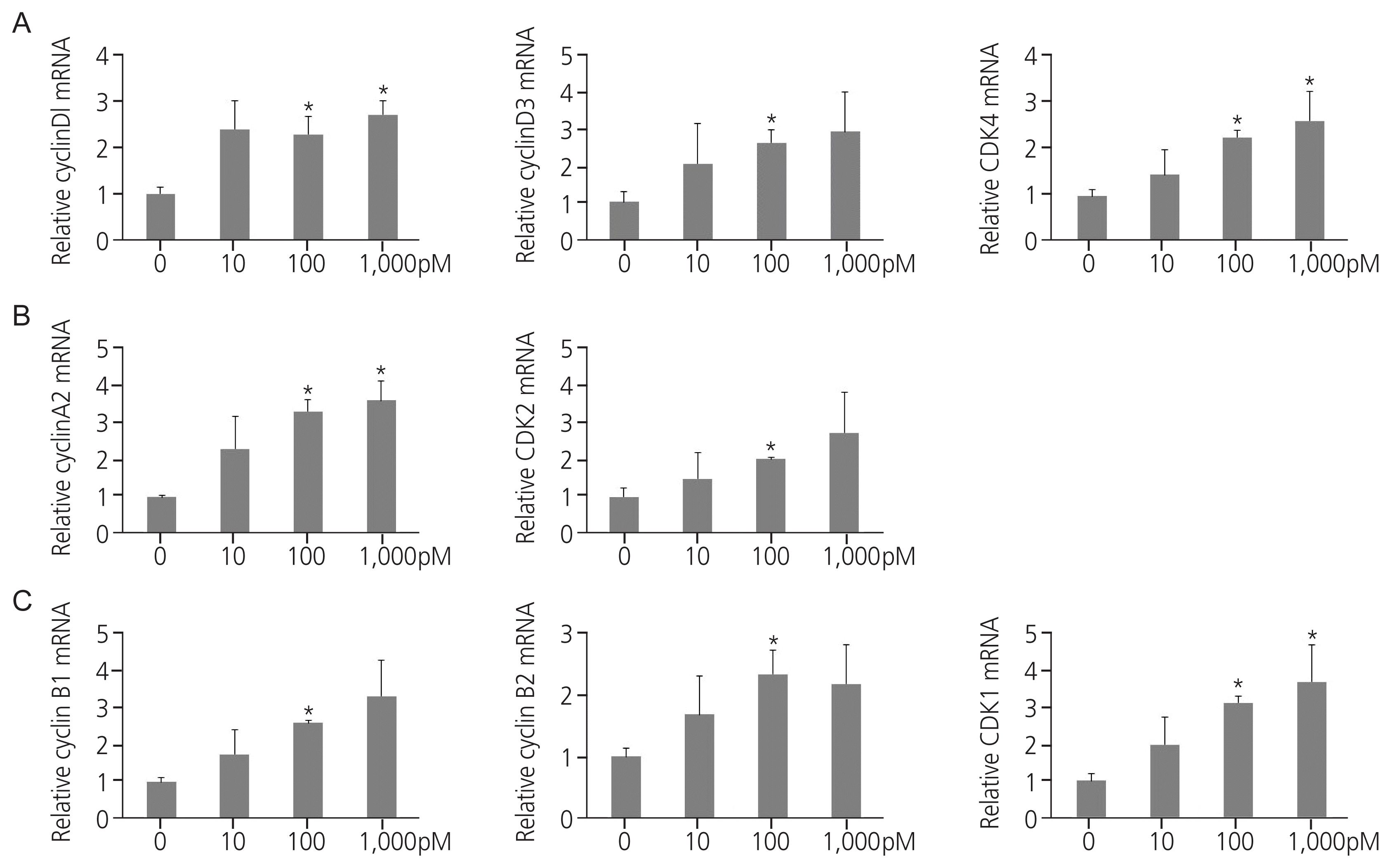
5. TPHP exposure enhances the G2/M phase of human Ishikawa endometrial cancer cells
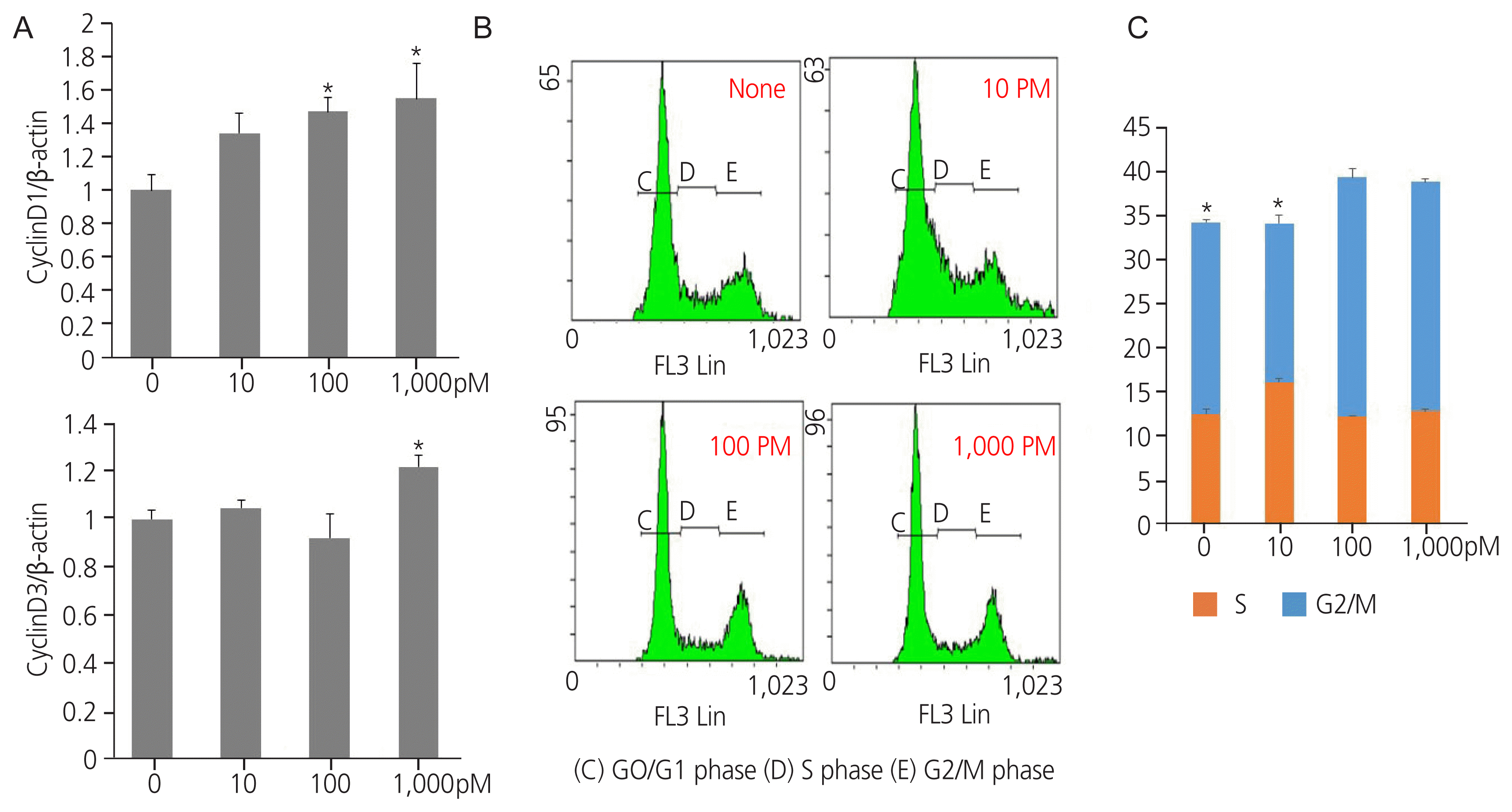
Endocrine disruptors stimulate cell cycle progression in cells by regulating cell cycle modulators. Exposure to TPHP elevated RNA levels of G1 phase and G2/M checkpoint markers (cyclin D1, D3, and CDK4) compared with the vehicle (Fig. 3A). In addition, TPHP exposure increased the levels of G2 phase markers (cyclin A2 and CDK2) (Fig. 3B) and M phase markers (cyclin B1, B2, and CDK1) in human Ishikawa endometrial cancer cells compared to the vehicle (Fig. 3C). Western blotting showed increased expression of cyclins D1, D3, and CDK4 (Fig. 4A). Collectively, the cyclin D/CDK4-6, cyclin A/CDK2, and cyclin B/CDK1 complexes promote progression in G1, progression in S, transition to G2, and G2/M transition, respectively, allowing entry into mitosis.
To validate the abovementioned observations, the proportion of cells in the G1, S, and G2/M phases of the cell cycle was estimated using flow cytometry (Fig. 4B, C).
Discussion
We showed that TPHP exposure activates the ER α/NF-κB/cyclin D axis, which may promote cell cycle progression and proliferation in human Ishikawa endometrial cancer cells. Previous cell-based transactivation studies have shown that TPHP upregulates the transcriptional activity of several nuclear receptors, including ER α [22]. In addition to the transcriptional activity of ER α, our study showed that TPHP increased ER α protein expression in human Ishikawa endometrial cancer cells. In the human endometrium, ER α expression is a critical driver of cell proliferation; however, the precise underlying molecular involvement remains to be fully elucidated. In previous studies, TPHP exposure altered DNA methylation patterns, and methylation of the ER α promoter region was correlated with ER α expression levels in breast cancer [23]. Thus, TPHP might promote hypomethylation of the ER α promoter region to increase ERα expression in human Ishikawa endometrial cancer cells.
In contrast to ER α expression, TPHP exposure reduced ER β protein expression in human Ishikawa endometrial cancer cells in the present study. The agonistic/antagonist activity of TPHP on ER β expression is controversial, and while TPHP enhanced the transcriptional activity of ER β in one study [22], it decreased the transcriptional activity at higher concentrations (1 and 3 μM) [24]. Hydroxylated TPHP metabolites are known to have definite ER β antagonist activity [24]. In the case of endometriosis, ER β knockdown significantly increased ER α mRNA and protein levels in endometriotic stromal cells [25]. We speculate that TPHP has an antagonistic effect on ER β and reduces its expression, enhancing ER α expression to stimulate ER α-mediated proliferation of human Ishikawa endometrial cancer cells.
Along with ER α, PR-B expression in human Ishikawa endometrial cancer cells was increased by exposure to 10–1,000 pmol TPHP. The effects of EDCs on PRs have been studied less than those on other nuclear receptors [26]. Although this is the first report to show that TPHP can alter PRs in human Ishikawa endometrial cancer cells, further studies are needed to explain the pathogenic role of PR in TPHP-induced endometrial dysfunction.
In the present study, TPHP exposure activated ER α function through the PI3K/AKT, p38, and MAPK/ERK signaling pathways in human Ishikawa endometrial cancer cells. Activation of the PI3K/AKT axis stimulates ER α function in ER α-positive breast cancer [27], and activated ERK signaling stimulates ER α activity in vascular smooth muscle cells and endothelial cells [28]. Thus, TPHP exposure may stimulate ER α activity in human Ishikawa endometrial cancer cells through the activation of the PI3K/AKT and ERK signaling pathways.
Cyclins are a family of proteins that play a role in cell cycle progression by activating CDKs. Some studies have reported on endocrine disruptors and cell cycle alterations. Perfluorooctanoic acid exposure promotes human breast epithelial cell proliferation by accelerating the G0/G1 to S phase transition of the cell cycle and increasing cyclin D1 and CDK4/6 levels [29]. Triclosan and octylphenol alter the expression of cyclin D1 in MCF-7 human breast cancer cells, which is linked to the G1/S transition of the cell cycle, leading to cell proliferation. These effects were also observed in vivo and were reversed by an ER antagonist, indicating that EDC-induced activity is mediated by an ER-dependent signaling pathway [30].
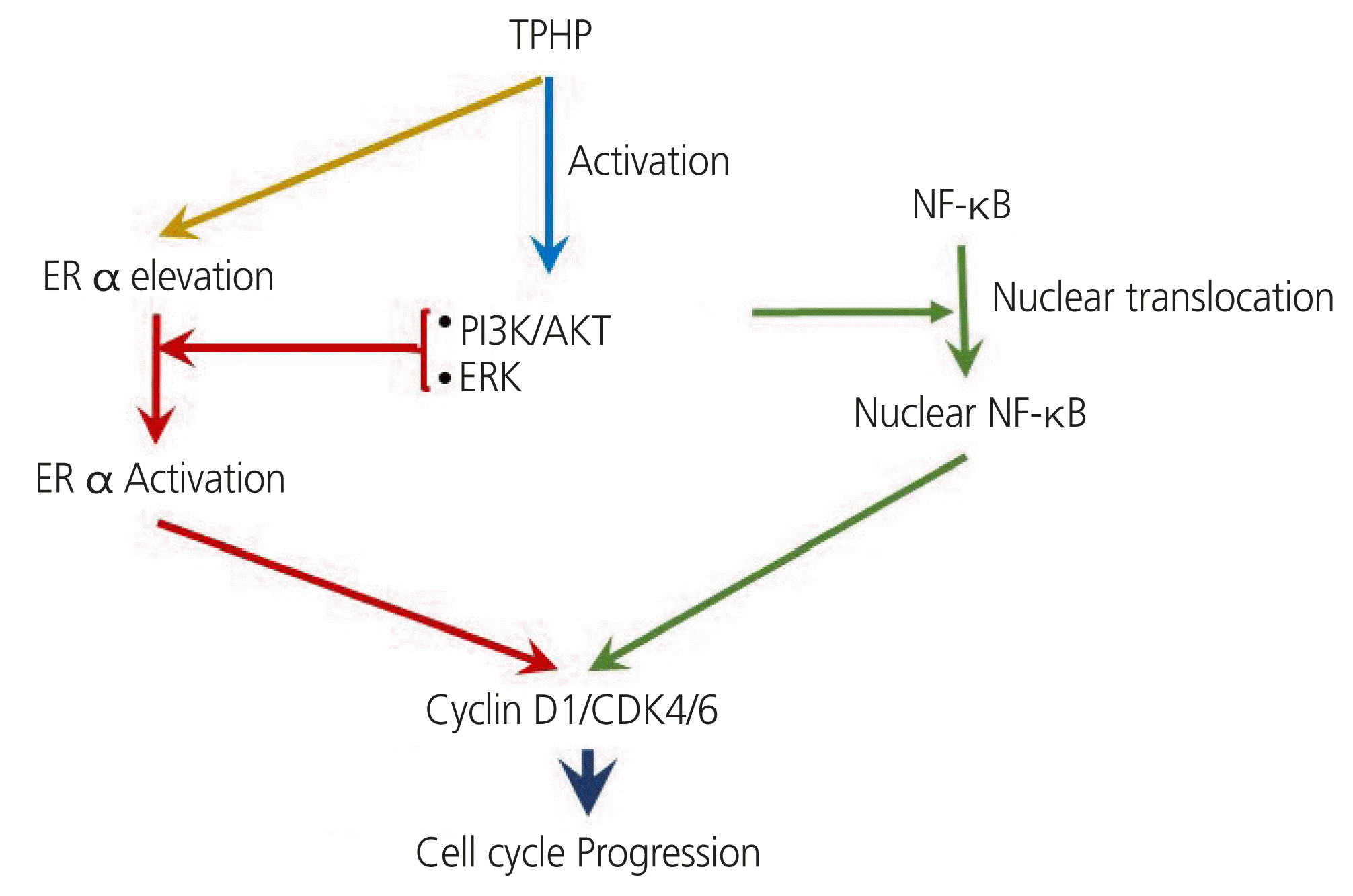
D cyclins are regarded as links between the cell environment and the core cell cycle machinery, and cyclin D1 is a direct transcriptional target of ER α [31]. Cyclin D1 is a positive regulator of ER α-mediated transcription and enhances the transcription of estrogen response element-responsive genes [32]. Cyclin D1 and CDK4/6 have ER α-dependent divergent functions in breast cancer progression [33]. The effects of TPHP exposure for 48 hours on the cell cycle regulatory proteins cyclin D1 and D3 are shown in Fig. 5. ER α expression is increased after TPHP treatment (Fig. 1), which may suggest that TPHP effects are mediated through an ER α-dependent signaling pathway. Therefore, activated ER α directly enhances the expression levels of cyclin D1 and CDK4/6 to stimulate cell cycle progression in human Ishikawa endometrial cancer cells.
Along with ER α, TPHP exposure increases nuclear NF-κB levels in human Ishikawa endometrial cancer cells. Interestingly, NF-κB regulates ER α transcription in the human heart and controls cell growth and differentiation through the activation of cyclin D1 expression [34]. Moreover, the PI3K/AKT axis is known to regulate NF-κB function, as NF-κB activity is essential for PI3K- and AKT-induced oncogenic transformation [35]. Thus, TPHP exposure activated PI3K/AKT to stimulate the nuclear translocation of NF-κB to stimulate the cyclin D1/CDK4/6 axis in human Ishikawa endometrial cancer cells.
Based on our results, we propose that (1) exposure to TPHP elevates levels of ER α, (2) activates PI3K/AKT and ERK kinase signaling, (3) increases the nuclear translocation of NF-kB, and (4) activates ER α and nuclear NF-κB stimulate expression of cyclin D1/CDK4/6, which may contribute to enhanced proliferation in human Ishikawa endometrial cancer cells (Fig. 5).
Our study had some limitations. As this is the first study to evaluate the effects of TPHP on human endometrium-derived cells, we focused only on TPHP. DPHP is the primary metabolite of TPHP, and the differences in the in vivo and in vitro effects of these two compounds remain to be elucidated.
Another consideration is that although the human Ishikawa endometrial cancer cell line is a standard model for the study of hormonally regulated events in the human endometrial epithelium, changes in heterogeneous cell populations during long-term culture have been reported [36]. To overcome these limitations, further studies are necessary to investigate divergent chemicals, including primary metabolites, in normal endometrial stromal cells and epithelial cells.
A final consideration is that we used very low doses of TPHP in this study (pmol to nmol range). In Chinese men and women, the concentration of TPHP in the blood was approximately 1.21 ng/mL [37]. TPHP has been detected in the human placenta at a concentration of 215 ng/g lw [38]. Many in vitro studies reporting the toxicological effects of TPHP have used concentrations in the μM range, which are much higher than those used in this study (1 nM corresponds to 1.305 ng/well for TPHP) [39]. As many EDCs show adverse effects at low-dose exposure with non-linear dose responses, we used very low, non-toxic levels to investigate the effect in this cell line, and all of the results were not exactly dose-dependent.
The results of this study demonstrate that TPHP may stimulate human Ishikawa endometrial carcinoma cell proliferation by inducing cyclin D1, which leads to S and G2/M cycle progression via activation of ER α and NF-κB mediated by the PI3K/AKT and MAPK/ERK1/2 signaling pathways (Fig. 5). The results of our study provide new information on the molecular effects of TPHP in a human Ishikawa endometrial cancer cell line.
 XML Download
XML Download