

 PDF
PDF Citation
Citation Print
Print



INTRODUCTION
General anesthesia and sleep have long been discussed in the neurobiological context owing to their commonalities such as unconsciousness, immobility, non-responsiveness to external stimuli, and lack of memory upon returning to consciousness. In 1855, it was hypothesized that a common mechanism might be involved in general anesthesia and normal deep sleep [1]. However, since the exact mechanisms of anesthesia and sleep are yet to be completely understood, the relationship between the two phenomena remains uncertain. This review discusses the current understanding of neurobiological mechanisms underlying sleep and anesthesia and explores potential clinical implications. Based on the similarities and differences between these seemingly similar states, we sought to gain a better insight into anesthesiology and sleep medicine.
DEFINITION OF GENERAL ANESTHESIA AND SLEEP
The American Society of Anesthesiologists defined general anesthesia as “drug-induced loss of consciousness during which patients are not arousable, even by painful stimulation” [2]. This condition is also accompanied by amnesia, akinesia, and the stability of life-sustaining physiological systems [3]. Sleep is characterized by immobility and a reduced level of responsiveness to the environment, unlike the awake state, and differs from hibernation or anesthesia in that it is quickly reversible [4]. Sleep is often regarded as a passive state; however, it is a dynamic process that is regulated by complex mechanisms rather than a state of inactivity. Sleep states are categorized into the following two distinct phases: rapid eye movement (REM) and non-rapid eye movement (NREM) sleep. REM sleep is accompanied by bursts of rapid eye movements, irregular breathing and heart rate, dreams, and muscular atonia, whereas NREM sleep is characterized by slow eye movement, decreased muscle tone, body temperature, and heart rate [5].
ELECTROENCEPHALOGRAPHIC FINDINGS IN GENERAL ANESTHESIA AND SLEEP
Electroencephalograms (EEG) reveal distinct patterns during general anesthesia. As the depth of anesthesia increases, the EEG pattern changes from paradoxical excitation, phases 1 and 2, followed by burst suppression and isoelectric traces [3]. During the induction period, EEG shows an increase in beta activity (13–25 Hz), implying a paradoxically excited brain status. Under lighter phases anesthesia (phase 1), EEG beta activity decreases while alpha (8–12 Hz) and delta (0.5–4 Hz) activities increase. An intermediate state (phase 2) is characterized by increased alpha and delta activity in the anterior brain, called anteriorization. The EEG of phase 2 anesthesia is similar to that of stage 3 NREM sleep. Under more profound anesthetic state (phase 3), a critical feature on EEG is intermittent high-power range oscillations (bursts) in alternation with isoelectricity (suppression), which is known as burst suppression [6–9]. This distinct feature is mainly observed when the brain is inactivated, such as during hypothermia [10–12], deep general anesthesia [3,13], and coma [14]. Surgical procedures are usually conducted in phases 2 and 3.
EEG during sleep also shows distinct stages. In NREM stage 1, the EEG frequency decreases, and strong alpha and theta activities are observed. In NREM stages 2 and 3, the amplitude of EEG increases, and the frequency reduces further, which are characterized by both sleep spindles (7–14 Hz) and K-complexes. In NREM stage 4, brain waves show high-amplitude and low-frequency rhythms. In particular, large-amplitude delta waves (0.5–4 Hz) are characteristic of NREM sleep stage 4, also known as slow-wave sleep. During REM sleep, EEG shows high-frequency and low-amplitude rhythms, and theta (7–9 Hz) activity is prominently observed [15]. More information can be found in the review by Brown et al. [3].
REGULATORY MECHANISMS FOR SLEEP AND WAKEFULNESS IN THE BRAIN
Arousal pathway
Activation of wake-promoting systems causes arousal in an organism and prevents it from falling asleep. During states of wakefulness, low-amplitude oscillations at 20–60 Hz were found in the cortical EEG, and an electromyogram (EMG) shows irregular muscle activities [16–18]. According to Moruzzi and Magoun [19], the waking state is maintained by an ascending flow of arousal signals emanating from the brainstem reticular formation. The ascending reticular activating system (ARAS) is generally located in the pons and midbrain. In addition to ARAS, many other wake-promoting areas were identified [20,21]. Recent advances in neuroscientific techniques have enabled the identification of intracerebral nuclei and their neurotransmitter-specific cell types which maintain or promote wakefulness. Optogenetics and chemogenetics have revolutionized the anatomical and physiological understanding of sleep and wakefulness. Optogenetics is a cutting-edge neurotechnology that uses light-sensitive proteins, such as channelrhodopsin, and illumination to control the activity of neurons in a cell-type- and region-specific manner with high temporal precision. Neurons induce or suppress action potentials when light-sensitive ion channels or pumps are stimulated optically [22]. Similarly, chemogenetics employs mutated muscarinic receptors, such as hM3Dq and hM4Di, which are responsive to the exogenous chemical clozapine-N-oxide (CNO) but not to the endogenous ligand acetylcholine. After hM3Dq or hM4Di is expressed in a specific group of cells, the specific neurons can be excited or inhibited selectively by CNO administration. Wake-promoting cell groups include noradrenergic cells in the locus ceruleus (LC) [23], 5-HT-synthesizing (serotonergic) cells in the dorsal raphe nuclei (DRN) [24], acetylcholine-synthesizing (cholinergic) cells [25–28], glutamatergic, and GABAergic cells [25,27,29] in the pedunculopontine tegmentum (PPT) and laterodorsal tegmentum (LDT), and DA-synthesizing (dopaminergic) cells in the ventral tegmental area (VTA) [30]. The parabrachial nucleus (PB) is also a strong wake-promoting area that is active during hypercarbia, pain, cold, and nausea [31]. In the forebrain, some nuclei promote wakefulness in conjunction with ARAS [16], including histaminergic cells in the tuberomammillary nuclei (TMN) [32], hypocretinergic (also known as orexinergic) cells in the lateral hypothalamus (LH) [33–36], cholinergic and parvalbumin-containing cells in the basal forebrain (BF) [37], and glutamatergic neurons in the PB [31]. Activation of these systems spontaneously or experimentally leads to a pattern of cortical activation required to maintain wakefulness. However, it remains unclear whether all these cell populations are simultaneously necessary to generate and maintain wakefulness.
Sleep-promoting pathway
First, the ventrolateral preoptic area (VLPO) was discovered as a sleep-promoting area. GABAergic and galaninergic neurons in the VLPO project to wake-promoting brain regions, such as the TMN, DRN, LC, PB, hypocretinergic neurons, and cholinergic neurons of the BF. Other areas were also found to have sleep-active neurons. The parafacial zone in the rostral medulla, which is dorsolateral to the origin of the facial nerve, contains GABAergic/glycinergic neurons. These neurons are active during NREM sleep, and their selective activation causes longer NREM sleep and higher EEG delta rhythm [38]. In the cortex, most neurons are wake-active, but neurons containing nitric oxide synthase (nNOS) in the deep layer are sleep-active [39]. These neurons fire in response to homeostatic sleep pressure and release GABA and nitric oxide, slowing down cortical rhythms. Additionally, two sets of neurons are essential for REM sleep. One is cholinergic neurons in the PPT/LDT area, which are active during REM sleep and are known as “REM-ON” cells [40]. The other is glutamatergic neurons in the sublaterodorsal nucleus, which have been discovered more recently as REM-sleep generators [41].
Flip-flop switch model
Interestingly, sleep-promoting VLPO and wake-promoting regions are connected by bidirectional inhibitory projections. This reciprocal inhibitory neural circuitry is similar to flip-flop circuitry in electronics, which enables rapid transitions between sleep and wake states and keeps the states in a stable manner. Thus, a model mechanism for sleep-wake control between VLPOs and arousal-promoting regions was proposed and named the “flip-flop switch” model [42]. Activating VLPO neurons promotes sleep by inhibiting arousal-promoting areas such as LC, TMN, and DRN neurons, whereas activating LC, TMN, and DRN neurons promote wakefulness by inactivating VLPO neurons. As a result of solid inhibition between these two sides of the switch, the system rapidly shift from one state to the other and stabilizes one state against the other. Therefore, balancing arousal-promoting and sleep-promoting circuitries is critical for determining a vigilance state.
Homeostatic sleep regulation
In general, prolonged wakefulness is followed by longer and deeper sleep. Homeostatic sleep response is characterized by a higher sleep propensity and longer sleep time after sleep deprivation (SD) [43]. Adenosine is a neurochemical substrate of sleep propensity and sleep-inducing factor that increases in proportion to prior wakefulness [44]. In contrast, slow wave activity (SWA) is considered an electrophysiological measure of sleep propensity or the tendency toward sleep and can be measured by the power of cortical delta waves (0.5–4 Hz). Homeostatic sleep response occurs during recovery sleep after SS and is characterized by increased SWA. Additionally, slow-wave energy (SWE), the time integral of SWA during NREM sleep, also represents the homeostatic sleep response.
ANESTHETIC AGENTS: GABAERGIC VS. NON-GABAERGIC
General anesthetics increase inhibitory neurotransmission and decrease excitatory neurotransmission in the central nervous system, ultimately leading to a suppressed state of the brain. However, the mechanisms of action of general anesthetics are not yet well established. In addition, the representative states of general anesthesia, including unconsciousness, amnesia, analgesia, and akinesia, are mediated by different receptors and brain regions. Most general anesthetics act on neurotransmitter-gated ion channels. It has been found that among the various ion channels, the γ-aminobutyric acid type A (GABAA) receptor plays a significant role as a functional site for general anesthesia [45–49]. GABA is the primary inhibitory neurotransmitter in the mammalian central nervous system and its inhibition is mediated mainly through chloride-permeable chloride-gated ion channels called GABAA receptors. Activation of GABAA receptors triggers an inflow of chloride ions into the cell, causing cell membrane potential hyperpolarization and decreased neuronal excitability [50]. The GABAA receptor has an active site to which GABA and other drugs, such as bicuculline, can bind as a ligand along with multiple allosteric binding sites [51]. Various anesthetics, including benzodiazepines, propofol, neuroactive steroids, barbiturates, and inhaled anesthetics, can modulate receptor activity by binding to the different allosteric binding sites [52]. Non-GABAergic agents such as ketamine [53], nitrous oxide [54], and xenon [55] rarely interact with GABAA receptors. The anesthetic effects are mediated by non-competitive antagonism of the N-methyl-D-aspartic acid (NMDA) receptor, resulting in decreased excitatory neurotransmission. Especially, ketamine interacts with the opioid, monoaminergic, cholinergic, purinergic, and adrenoreceptor systems [56,57].
THE ACTION OF ANESTHETICS BLOCKING THE AROUSAL PATHWAY
Tuberomammillary nucleus
The TMN is a critical wake-promoting nucleus and a target for anesthesia-induced sedation [58]. In particular, after sedation induced by GABAergic anesthetics such as propofol, muscimol, and pentobarbital, the expression pattern of the c-Fos gene, a marker of neuronal activity, appears similar to that of NREM sleep. Expression of c-Fos, a marker for neuronal activation, decreased in TMN while increased in the VLPO, suggesting that GABAergic agents activate the VLPO and suppress the TMN. Furthermore, muscimol directly injected into the TMN increased the loss of righting response (LORR) time, a behavioral measure of the degree of sedation, in a dose-dependent manner, whereas GABAA receptor antagonist, gabazine reduced the LORR [58,59]. These findings suggest that GABAergic anesthetics inhibit neuronal activity in the TMN, resulting in reduced release of histamine and loss of consciousness [60,61].
Locus ceruleus
LC noradrenergic neurons have strong wake-promoting effects, and pre-and post-synaptic mechanisms contribute to the suppression of LC neuronal activity by general anesthesia. Neuronal loss in the LC of the zebrafish model resulted in faster induction and slower emergence from general anesthesia [62]. In addition, chemogenetic activation of LC noradrenergic neurons reduces delta power during isoflurane anesthesia and shortens its emergence time. Conversely, adrenoreceptor antagonists lenghthens the duration of anesthesia [63].
Hypocretinergic neurons
Hypocretinergic neurons also mediate anesthetic effects by decreasing their wake-promoting functions. Evidence suggests that isoflurane and sevoflurane decrease c-Fos expression in hypocretinergic neurons, also known as orexinergic neurons, implying that their activity is suppressed [64]. More recently, these neurons in the perifornical area of the lateral hypothalamus have demonstrated influencing the maintenance and emergence from isoflurane and desflurane anesthesia [65]. In addition, these neurons also affect induction via projections to the paraventricular nucleus.
DOES ANESTHESIA HAVE SLEEP-LIKE EFFECTS?
To date, many researchers have concluded that sleep and anesthesia are distinct states, but they share several similarities [66,67]. Although it is already known that anesthetics can induce sedation by acting on arousal neural circuits [58], no clear evidence showed that sleep-promoting nuclei cause sedation until volatile anesthetics, such as isoflurane or halothane, was examined for the activity of the sleep-promoting VLPO region by measuring the expression level of c-Fos [68]. As a result of volatile anesthetics administered during the dark period, an active period for the rodents, c-Fos-positive neurons increased in a dose-dependent manner, similar to the activation level of VLPO neurons during spontaneous sleep during the light period.
CAN ANESTHESIA REDUCE SLEEP PROPENSITY?
Sleep characteristics are homeostatically regulated based on the length and depth of previous sleep. Therefore, it is reasonable to question whether anesthesia can function in the same manner as sleep homeostasis. General anesthesia with a slow-wave or isoelectric EEG after sleep deprivation can result in a blunted SWA rebound, implying that anesthesia may have a similar effect to NREM sleep [69]. According to a study comparing the cumulative SWE of the group that received isoflurane for 1 h immediately after sleep deprivation for 4 h against the group that did not receive isoflurane, the slope of rebound sleep was flattened in the group treated with isoflurane [69]. This phenomenon can have two inferences. First, anesthesia may have caused an abnormal brain condition suppressing SWA and the abnormality sustained during recovery sleep. Another possibility is that anesthesia may have resolved the accumulated sleep pressure by an unknown mechanism. However, because both the high and low doses of anesthetic agents reduced SWA during recovery sleep and only a low dose could generate slow waves, anesthesia SWA may not be necessary for the sleep-like effect. Instead, there may be an unknown mechanism that mediates the sleep-like effects of general anesthesia. Sevoflurane may have similar but different effects on NREM and REM sleep. Six-hour sevoflurane anesthesia following 12-h sleep deprivation reduced the homeostatic response in NREM sleep but not in REM sleep [70]. Sevoflurane treatment after sleep deprivation reduced rebound sleep compared with no sevoflurane treatment, suggesting that homeostatic sleep pressure was partly resolved by sevoflurane anesthesia.
CAN ANESTHESIA AMELIORATE SD-INDUCED COGNITIVE DYSFUNCTION?
Sleep deprivation is commonly associated with memory impairment, one of the most prevalent symptoms. Therefore, it is unclear whether sedation with anesthetics also reduces SD-induced memory impairments. Dexmedetomidine, a selective α2-adrenergic receptor agonist, appears to counteract memory impairment induced by sleep deprivation [71]. Chronically sleep-deprived mice simultaneously treated with dexmedetomidine showed decreased memory impairment. Simultaneously, they showed lower levels of inflammatory cytokines, such as tumor necrosis factor-α (TNF-α) and interleukin (IL)-6, and increased levels of neuroprotective signaling molecules, such as brain-derived neurotrophic factor (BDNF) and tyrosine kinase B (TrkB). Dexmedetomidine reduces detrimental responses caused by sleep deprivation, potentially resembling sleep effects.
DOES SLEEP PRESSURE POTENTIATE THE EFFECTS OF THE ANESTHETICS?
We have reviewed that anesthesia may have sleep-like effects. In contrast, whether sleepiness or sleep deprivation can affect anesthesia is a clinically important question. Twenty-four-hour sleep-deprived rats showed a shorter time taken for loss of righting reflex and a longer time to recovery when anesthetized with propofol and isoflurane, implying that they presented faster induction and slower emergence from anesthesia [70,72]. These data support the hypothesis that increased sleep pressure enhances the effects of propofol and isoflurane. Given that preoperative sleep disturbance is associated with an increased risk of postoperative delirium [73] and that increased sleep pressure may potentiate the efficacy of anesthetic agents [72], it is clinically relevant to determine whether dose reduction could be considered for patients with either a pre-existing medical or psychiatric condition that hinders sleep chronically, or acute preoperative sleep disruption, or both. These findings suggest that sleep propensity could contribute to the response to anesthetics and that sleep quantity and quality may contribute to individual differences in responses to anesthetics.
CHALLENGES TO THE “SHARED CIRCUIT HYPOTHESIS”
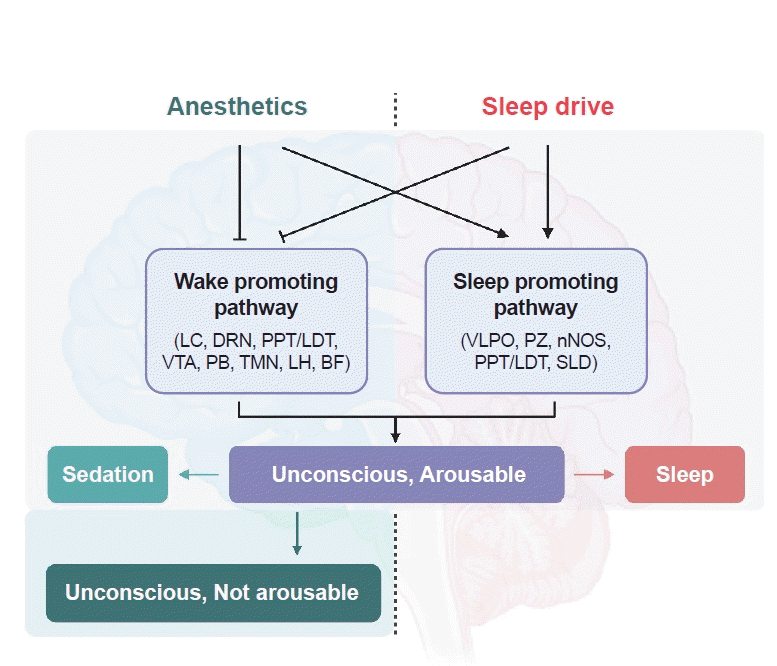
The “shared circuit hypothesis” proposes that the activation of sleep-promoting neurons is necessary or sufficient for attaining unarousable unconsciousness or general anesthesia [33]. This hypothesis has persisted for a long time but has recently been challenged. Ablation of the VLPO, a major sleep-promoting area, could increase the sensitivity to isoflurane. However, VLPO-ablated rats did not lose the ability to achieve deep isoflurane anesthesia [74]. In addition, a recent study by Vanini et al. showed that selective activation of VLPO using chemogenetics did not facilitate anesthesia. Briefly, they selectively activated GABAergic neurons in the VLPO by activating the hM3Dq receptor with CNO and found no difference in the time taken for loss and recovery of consciousness by isoflurane anesthesia. These findings disprove that VLPO is not necessary or sufficient to induce general anesthesia. Nevertheless, it should be noted that there are multiple levels of anesthetic action (Fig. 1). Anesthetic agents can cause unconscious but arousable states by acting on sleep-promoting areas, such as the VLPO, LH, LC, and TMN. For a deeper level of anesthesia, such as the unconscious and non-arousable state, anesthetics should act directly on the neural substrate of consciousness [75].
CONCLUSION
Recent advances in understanding the neurobiology of sleep, wakefulness, and anesthesia have elucidated the bidirectional relationship between sleep and anesthesia. Neural circuits of sleep-wake regulation are affected by anesthetics, and, in turn, anesthesia is affected by sleep propensity. Although sleep and anesthesia do not occur simply by the same mechanism or neural circuitry, their interaction might be important and meaningful for future investigations. Systematic research on the relationship between sleep disturbance and anesthesia is warranted.
 XML Download
XML Download