

 PDF
PDF Citation
Citation Print
Print


INTRODUCTION
The global prevalence of type 2 diabetes remains high, with the number of people projected to reach 693 million in 2045 [1]. Diabetes has become a serious burden on patients’ families and society due to its serious complications and poor prognosis. Diabetes can cause persistent damage to systemic organs [2], and cardiovascular complications are the leading cause of death in patients with diabetes, especially those with type 2 diabetes [3]. Diabetic cardiomyopathy, as one of the independent and important cardiovascular complications of diabetes, is also an important cause of death and disability in patients with type 2 diabetes [4-7]. Diabetic cardiomyopathy, which can lead to poor prognosis, such as heart failure and acute cardiovascular events [8,9], has become a major public health problem worldwide in the chronic disease management of diabetes.
The key link in the occurrence and development of diabetic cardiomyopathy is myocardial fibrosis. Myocardial fibrosis is pathological remodeling of the myocardium characterized by myocardial cell loss, excessive deposition of myocardial extracellular matrix, and changes in the phenotype of cardiac fibroblasts [10]. Various injury factors can promote the imbalance of myocardial cell homeostasis to induce myocardial cell apoptosis, resulting in pathological myocardial remodeling, reduced ventricular wall motility, and heart failure [11]. The pathogenesis of diabetic myocardial fibrosis is not very clear at present, and there is currently a lack of clear and effective clinical interventions for diabetic myocardial fibrosis.
Apoptosis, the most common way of programmed cell death, plays an important role in the pathological outcome of many diseases. Studies have shown that high glucose levels caused by diabetes can produce glucotoxicity and lead to cardiomyocyte apoptosis, and high glucose can cause mitochondrial damage and ROS accumulation in cardiomyocytes. Finally, the apoptosis program is initiated, which can lead to diabetic cardiomyopathy [12-14]. Moderate autophagy is critical for cellular homeostasis under stress, and glucotoxicity from high glucose levels inhibits cardiomyocyte autophagy [14-16]. Impairment of autophagy can lead to cardiomyocyte apoptosis, excessive repair of fibrosis, and pathological remodeling [17]. However, the role of autophagy in cardiomyocyte apoptosis and myocardial fibrosis induced by type 2 diabetes is still unclear and needs further study.
As an important pathway mediating autophagy, the phosphatidylinositol 3-kinase (PI3K)/AKT (AKTV-murine thymoma viral oncogene/protein kinase B) signaling pathway can regulate cell survival and homeostasis, and is an important target of cardiovascular disease [18,19]. Excessive activation of the PI3K/AKT signaling pathway induced by a high-glucose and high-fat environment can lead to abnormal organ function. A recent study found that the PI3K/AKT pathway was significantly up-regulated in the myocardial tissue of rats with diabetic cardiomyopathy, accompanied by impaired autophagosome formation and defective autophagy [20]. Previous studies have found that inhibition of PI3K/AKT can activate autophagy, thereby alleviating fibrosis and cell damage [21,22]. In the process of myocardial fibrosis, inhibiting the excessive activation of PI3K/AKT can inhibit the progression of myocardial fibrosis [23]. In addition, studies have shown that in the myocardial tissue of rats with diabetic cardiomyopathy, inhibiting the activation of the PI3K/AKT signaling pathway can reduce oxidative stress, thereby improving impaired cardiac function and reducing pathological changes such as fibrosis [24]. However, the role of PI3K/AKT-mediated autophagy in the occurrence and development of diabetic cardiomyopathy is still unclear.
Sulfur dioxide (SO2) is generally known as “a toxic gas.” But in recent years, it has been found that SO2 is also an important endogenous gas signal molecule like NO and CO. SO2 can be endogenously generated in vivo by an enzymatic reaction catalyzed by aspartate aminotransferase (AAT) in the metabolic pathway of sulfur-containing amino acids. It has been found to play an important role in cardiovascular homeostasis and collagen remodeling [25]. Studies have shown that the expression of SO2-producing enzymes in the myocardial tissue of diabetic rats is low [26], and SO2 can inhibit the proliferation and migration of fibroblasts and the progression of myocardial fibrosis [27]. However, it is unclear whether SO2 can antagonize myocardial fibrosis in diabetic rats and its internal regulation mechanism. The effect of SO2 on autophagy and apoptosis of myocardial cells in diabetic rats is also unclear.
Therefore, this study intends to construct the diabetic rat model and give exogenous SO2 donors to observe the effect of gas signaling molecule SO2 on myocardial cell apoptosis and myocardial fibrosis in rats with type 2 diabetes and to explore whether its mechanism is related to the regulation of autophagy by PI3K/AKT pathway.
Go to : 
METHODS
Materials
Streptozotocin (STZ), Na2SO3, NaHSO3, D-glucose, and HDX (serine racemase inhibitor) were purchased from Sigma-Aldrich (St. Louis, MO, USA). PI3K/AKT activator 740 Y-P was provided by MedChemExpress (Shanghai, China). PI3K (67071-1-Ig), AKT (10176-2-AP), Beclin1 (11306-1-AP), LC3 (14600-1-AP), Atg5 (10181-2-AP), Atg16L1 (19812-1-AP), P62 (66184-1-Ig), AAT1 (14886-1-AP), AAT2 (14800-1-AP), Caspase3 (19677-1-AP), Caspase9 (10380-1-AP), Bax (50599-2-Ig), Bcl2 (26593-1-AP), MMP3 (66338-1-Ig), MMP8 (17874-1-AP), MMP13 (18165-1-AP), CollagenIII (22734-1-AP), and GAPDH (10494-1-AP) primary antibodies and mouse/rabbit secondary antibodies were purchased from Proteintech (Wuhan, China). P-PI3K (4228S) and P-AKT (9614T) primary antibodies were offered by Cell Signaling Technology (Beverly, MA, USA). Gel configuration kits and BCA protein quantification kits were purchased from Biyuntian Institute of Biotechnology (Shanghai, China). ECL chemiluminescence kit was provided by New Cell and Molecular Biotech Co., Ltd. (Suzhou, China). AnnexinV-fluorescein isothiocyanate/propidium iodide (AV-FITC/PI) apoptosis detection kit was purchased from Keygene Biotech Co., Ltd. (Nanjing, China).
Animals
The animal experiments were performed in accordance with the guidelines for the welfare and use of laboratory animals and gained approval from the Animal Ethics Committee of the University of South China, according to the animal management regulations and specifications of the Laboratory Animal Center of University of South China. A total of 40 8-week-old male Sprague–Dawley (SD) rats were randomly assigned to four groups (n = 10). The type II diabetes rats model was established through the single intraperitoneal injection of STZ (35 mg/kg, Sigma-Aldrich; dissolved in citric acid-sodium citrate buffer, pH 4.5) combined with a diet high in fat and sugar for eight weeks before STZ intraperitoneal injection [28]. The Control group and the SO2 group were given an equal amount of citric acid-sodium citrate buffer through intraperitoneal injection. One week after the injection, fasting blood glucose levels of the rats > 16.7 mM indicated that the type II diabetes rat model was successfully established, and diabetic rats were continuously given a diet high in sugar and fat. Subsequently, the diabetic cardiomyopathy (DC) + SO2 and SO2 groups were given exogenous SO2 donor (Na2SO3/NaHSO3, 85 mg/kg/d, Sigma-Aldrich) intraperitoneally for eight weeks [29], and the Control group and the DC groups were given equal amounts of saline intraperitoneally (Fig. 1). The heart tissues were collected after eight weeks of SO2 intervention for follow-up assays, and the heart weight and the pre-execution weight were weighed to calculate the HW/BW value. 8-week-old male SD rats were purchased from HUNAN SJA Laboratory Animal Co. (Changsha, China).
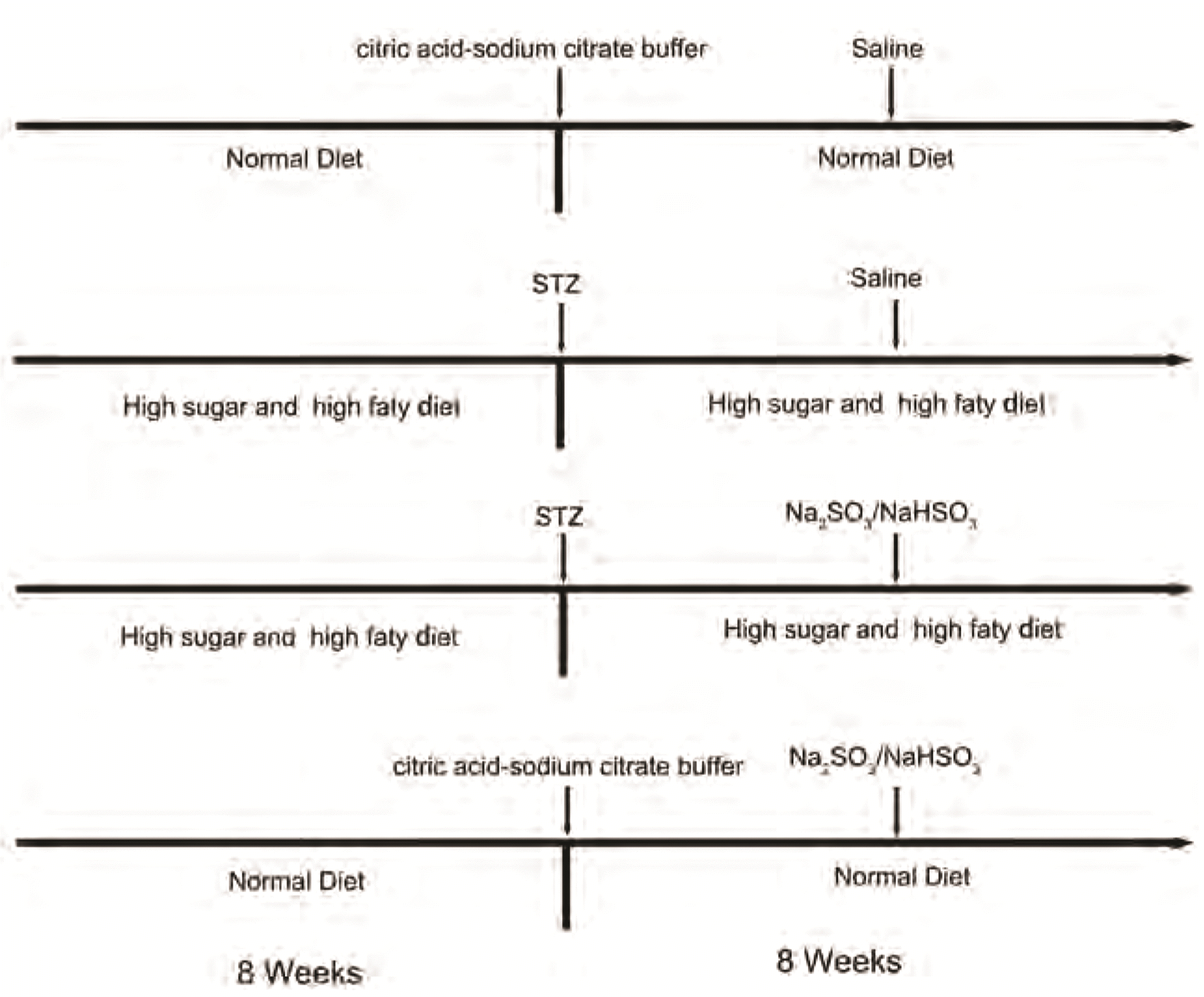 | Fig. 1Animal grouping and drug treatment.Sprague–Dawley (SD) rats were administered with 8 weeks of different dietary interventions followed by intraperitoneal injection of streptozotocin (STZ) or citric acid-sodium citrate buffer, and the dietary treatment was continued for 8 weeks after the successful establishment of the diabetic rats model, meanwhile intraperitoneal injection of exogenous sulfur dioxide (SO2) donor (Na2SO3/NaHSO3) or equivalent saline for 8 weeks. DC, diabetic cardiomyopathy.
|
Blood biochemistry analysis
After eight weeks of STZ injection, all rats were gavaged with 50% glucose solution for the glucose tolerance test, and tail vein blood specimens were collected at 30, 60, and 120 min to measure the blood glucose levels. Fasting blood glucose and fasting insulin levels were collected from the rats at the time of devotion and homeostasis model assessment of insulin resistance (HOMA-IR) values were obtained.
Echocardiography
After all rats were anesthetized with 4% chloral hydrate (400 mg/kg), the changes of left ventricular end-diastolic-dimension (LVEDD), left ventricular end-systolic dimension (LVESD), and left ventricular fractional shortening (LVFS) values were measured using M-mode ultrasound.
Histopathological staining
Myocardial tissues were fixed with 4% paraformaldehyde, paraffin-embedded after dehydration by gradient alcohol, made into 4 μm thin sections by microtome, and stained in accordance with the instructions of the Masson staining kit (Abiowell Biotech Co., Ltd., Changsha, Hunan), and sealed by treacle. The staining of the tissues was observed under a light microscope. The fixed and embedded myocardial tissues were made into 4um thin sections, and the corresponding operations were performed in accordance with the instructions of the TUNEL staining kit (Keygene Biotech Co., Ltd.). The staining was observed under a light microscope.
Transmission electron microscopy
The myocardial tissues were fixed by 2.5% glutaraldehyde and cut into 50–100 nm thin sections, and phosphoric acid rinsing solution (Beyotime Institute of Biotechnology, Shanghai, China) was used. After being immersed in 1% osmium tetroxide (Absin Biosciences, Inc., Shanghai, China), the tissues were fixed and rinsed with phosphoric acid, followed by gradient acetone immersion. After being dehydrated and dried, the tissues were stained with 3% uranyl acetate and lead nitrate for 10–20 min and rinsed with distilled water. The ultrastructure was observed under transmission electron microscopy.
Cell culture
H9c2 cardiomyocyte cell line was purchased from ATCC (Manassas, VA, USA) and incubated in DMEM medium supplemented with 5.5 mM D-glucose with 10% fetal bovine serum and 1% penicillin/streptomycin at 37°C and 5% CO2. The cells were inoculated in 6-well plates when the cell density was close to 1 × 106 and incubated with a medium supplemented with high D-glucose (33 mM). The cells were assigned to the Control group, the HG group, the HG + SO2 group, the HG + SO2 + serine racemase inhibitor (HDX) group, the HG + SO2 + 740 Y-P group, and the SO2 group in accordance with different treatments. The Control group and SO2 group were cultured with low D-glucose medium (5.5 mM), and the HG group and HG + SO2 group, and HG + SO2 + HDX group were treated with high D-glucose medium (33 mM) for 24 h at 37°C. The exogenous SO2 donor Na2SO3/NaHSO3 (50 μM, 3:1 mol rate) was introduced to the HG + SO2 group and HG + SO2 + HDX group for the 0.5-h pretreatment as described previously [30], and then co-incubated with high D-glucose medium. Besides, the HG + SO2 + HDX group was co-incubated with the endogenous SO2-produced enzyme inhibitor HDX (200 μM) [31]. In order to verify whether SO2 activates autophagy by inhibiting the PI3K/AKT pathway, the HG + SO2 + 740 Y-P group was incubated with PI3K/AKT activator 740 Y-P (30 μM) after the SO2 pretreatment, and all groups were incubated with high D-glucose medium (33 mM), except for the Control group and the SO2 control group.
Flow cytometry analysis
H9c2 cardiomyocyte apoptosis was detected using AV-FITC/PI Apoptosis Detection Kit (Keygene Biotech Co., Ltd.). In accordance with the manufacturer’s instructions, the cells were digested with 0.25% EDTA-free trypsin (Gibco, Grand Island, NY, USA) and washed twice in cold phosphate buffer saline (PBS). Resuspension was performed with 500 μl binding buffer supplemented with 5 μl FITC and 5 μl PI for 10 min in a dark room. Flow cytometry (fluorescence-activated cell sorting [FACS] Calibur cytometer; BD Biosciences, San Diego, CA, USA) was adopted to obtain the apoptosis rate. AV-FITC values were set as the horizontal axis, and PI values were set as the vertical axis. All experiments were performed in triplicate.
Immunofluorescence staining
Cells were fixed with 4% PFA for 40 min, permeabilized with 0.1% Triton X-100 for 5 min × 2, and blocked with 5% BSA for 1 h, incubated with rabbit anti-Beclin1 primary antibody (1:200) overnight at 4°C, then incubated with Cy3 (1:200)-conjugated secondary antibody for 1 h at room temperature and counterstained using DAPI. Fluorescence was analyzed using a fluorescence microscope (IX71; Olympus, Tokyo, Japan).
Real-time q-PCR
Total RNA was extracted from the rat myocardial tissues using TRIzol (Thermo Fisher Scientific, Rockford, IL, USA) and then transcribed into cDNA with the Prime-Script RT kit (Takara, Shiga, Japan). Real-time qPCR was performed with SYBR green (Takara) under the standard conditions of 95°C for 30 sec, 40 cycles of 95°C for 5 sec, and 60°C for 30 sec. Real-time qPCR was conducted under the standard conditions, GAPDH was used as an internal reference, and the relative expression levels of target genes were obtained using the 2-ΔΔCt method. The primer sequences are presented as follows:
Western blotting
The rat myocardial tissues and the cell precipitates were lysed through radioimmunoprecipitation assay (RIPA) lysis buffer containing cold protease inhibitor, the supernatant was collected after centrifugation, and the protein concentration of the supernatant was quantified and leveled using the BCA protein quantification kit based on the manufacturer’s guidelines. The samples were denatured after heating at 95°C for 10 min, separated in 10%–12% polyacrylamide electrophoresis gels, and electrophoretically transferred to a PVDF membrane (polyvinylidene fluoride membrane; Millipore Co., Bedford, MA, USA). The membranes were blocked with 5% skim milk at RT for 1 h and treated with primary antibody at 4°C overnight. The membranes were washed three times (5 min × 3) with tris-buffered saline (TBST) containing 20 and incubated with the corresponding species of horseradish peroxidase (HRP)-conjugated secondary antibody at RT for 1 h. After TBST washing (5 min × 3), the exposure shots were taken with ECL chemiluminescent solution, and image signals were obtained using the Bio-Rad XRS+ imaging system (Bio-Rad, Hercules, CA, USA). The relative data were analyzed using ImageJ software (NIH, Bethesda, MD, USA).
Statistical analysis
The data were expressed as mean ± standard error (SEM). The statistical analysis was performed using GraphPad Prism 9.0 software (GraphPad Software Inc., San Diego, CA, USA). The differences between group means were evaluated through one-way ANOVA Test. p < 0.05 was considered statistically significant.
Go to :
RESULTS
Test results of related biochemical markers of blood
Tables 1 and 2 list the results of the glucose tolerance test and the values of fasting blood glucose, insulin and HOMA-IR of the rats in the respective group, respectively. The fasting blood glucose, insulin levels and HOMA-IR values of the rats in the DC group and the DC + SO2 group increased significantly compared with those of the Control group (p < 0.05), which suggested that the rat model of type II diabetes was successfully established. There were significant abnormalities of blood glucose and insulin in both groups. The fasting blood glucose, insulin levels and related HOMA-IR values in the DC + SO2 group tended to decrease compared with those of the DC group, whereas the difference between the two groups did not achieve statistical significance (p > 0.05). The fasting blood glucose, insulin level and HOMA-IR value of the rats in the SO2 group did not change significantly compared with those of the Control group (p > 0.05).
Table 1
Test results of glucose tolerance test of the rats in the respective group
| Groups | Number | 0.5 h (mM) | 1 h (mM) | 2 h (mM) |
|---|---|---|---|---|
| Control | 9 | 5.57 ± 0.24 | 5.68 ± 0.37 | 4.27 ± 0.16 |
| DC | 7 | 31.36 ± 0.62* | 31.09 ± 0.40* | 27.59 ± 1.34* |
| DC + SO2 | 8 | 30.62 ± 1.53* | 30.63 ± 0.44* | 24.83 ± 1.12* |
| SO2 | 9 | 6.94 ± 0.49 | 5.82 ± 0.89 | 5.54 ± 0.10 |
![]()
Table 2
Comparison of values of fasting blood glucose, insulin and HOMA-IR of the rats in the respective group
| Groups | Number |
Fasting blood glucose (mM) |
Fasting insulin level (μlU/ml, 103) |
HOMA-IR |
|---|---|---|---|---|
| Control | 9 | 3.70 ± 0.32 | 25.00 ± 10.00 | 4.00 ± 0.80 |
| DC | 7 | 23.15 ± 1.49* | 436.67 ± 79.46* | 363.03 ± 68.61* |
| DC + SO2 | 8 | 22.32 ± 1.27* | 316.67 ± 56.25* | 312.48 ± 58.59* |
| SO2 | 9 | 3.60 ± 0.28 | 44.00 ± 5.48 | 7.00 ± 0.63 |
![]()
Changes in the expression of endogenous SO2-generating enzymes AAT1/2
Western blot was used to detect the changes in protein expression of SO2-generating enzyme AAT1/2 in the myocardial tissues of the rats in the respective group (Fig. 2A, C). As revealed by the results, compared with the Control group, the expression of AAT1/2 in the myocardial tissues of the rats in the DC group decreased significantly (p < 0.05), while the expression of AAT1/2 in the myocardial tissues of the rats in DC + SO2 group increased after the intervention of exogenous SO2 donor Na2SO3/NaHSO3 (p < 0.05). There was no statistically significant difference in the expression of AAT1/2 between the SO2 control group and the Control group.
 | Fig. 2Exogenous sulfur dioxide (SO2) can up-regulate the expression level of aspartate aminotransferase (AAT)1/2.(A, C) Western blot analysis of the expression changes of AAT1/2 in the myocardial tissue of type II diabetes rats. n = 3, *p < 0.05 vs. Control; #p < 0.05 vs. DC. (B, D) Western blot analysis of the expression changes of AAT1/2 in H9c2 cardiomyocytes; n = 3, *p < 0.05 vs. Control; #p < 0.05 vs. HG; $p < 0.05 vs. HG + SO2. Values are presented as mean ± SD. DC, diabetic cardiomyopathy; HG, high glucose.
|
Besides, similar changes were also identified in the cell experiments in vitro (Fig. 2B, D). AAT1/2 in H9c2 cardiomyocytes of the HG group had significantly lower expression than that in the Control group (p < 0.05), while the expression of AAT1/2 in the HG + SO2 group was significantly up-regulated as compared with that in the HG group (p < 0.05). After the cardiomyocytes of the HG + SO2 + HDX group were intervened with the endogenous SO2 inhibitor HDX, the expression of AAT1/2 was significantly lower than that of the HG + SO2 group (p < 0.05), thus revealing that the production of SO2 in the myocardial tissue and cardiomyocytes of diabetes rats was reduced based on high-glucose stimulation. Furthermore, exogenous SO2 donors could up-regulate the expression of SO2-producing enzyme AAT1/2 in the myocardial tissue and high-glucose-stimulated myocardial cells of diabetic rats.
Effects of SO2 on cardiac structure and cardiac function in diabetic rats
In this paper, we detected the cardiac function and structural changes of the respective rats by echocardiography (Table 3), and found that compared with the Control group, the LVFS value of the DC group decreased, while the LVFS value of the DC + SO2 group increased after SO2 intervention. However, the difference between the groups was not statistically significant. By comparing the changes of HW/BW of the rats in the respective group, as listed in Table 4, we found that the HW/BW of the DC group was significantly higher than that of the Control group (p < 0.05). However, after SO2 intervention, the HW/BW of the DC + SO2 group decreased; the difference was not statistically significant.
Table 3
Cardiac LVFS values of the rats in the respective group
![]()
Table 4
The HW, BW and HW/BW values of the rats in the respective group
| Groups | Number | BW (g) | HW (mg) | HW/BW (10–3) |
|---|---|---|---|---|
| Control | 9 | 390.00 ± 2.58 | 1090.00 ± 93.63 | 2.79 ± 0.17 |
| DC | 7 | 236.25 ± 19.70 | 810.50 ± 47.50 | 3.48 ± 0.09* |
| DC + SO2 | 8 | 285.83 ± 24.13 | 914.67 ± 85.39 | 3.20 ± 0.11 |
| SO2 | 9 | 400.00 ± 25.49 | 1017.20 ± 45.26 | 2.55 ± 0.09 |
![]()
The effect of SO2 on myocardial fibrosis in diabetic rats
As revealed by the result of Masson staining (Fig. 3A, B), compared with the control group, the blue-stained collagen fibers in the myocardial tissue of the diabetic rats in the DC group were significantly increased by about 4 times, and the myocardial fibers were disordered. However, after the intervention of exogenous SO2, the number of blue-stained collagen fibers in the myocardial tissue of rats decreased by about 1/2 compared with the DC group (p < 0.05), and the myocardial cells were arranged relatively neatly. Moreover, the expression changes of fibrosis-associated proteins (including Collagen III, MMP3, MMP8, and MMP13) were detected through Western blot (Fig. 3C, D). It was found that the expression of Collagen III, MMP3, MMP8, and MMP13 in the DC group was significantly higher than those in the Control group (p < 0.05). As revealed by the RT-qPCR results (Fig. 3E), the mRNA expression of Col3a1 in the DC group was about 3.8 times higher than that in the Control group; after the SO2 intervention, the changing trend of the above fibrosis-associated proteins was significantly reversed (p < 0.05). However, no statistically significant difference was found between the Control and SO2 groups. The above results indicated that there was obvious myocardial fibrosis in type II diabetes rats, and SO2 intervention could ameliorate myocardial fibrosis in diabetic rats.
 | Fig. 3Sulfur dioxide (SO2) can reduce the formation of myocardial collagen fibers in type II diabetic rats.(A) Masson staining results of myocardial tissue of the rats in the Control, DC, DC + SO2, and SO2 groups. 10 × 40 magnification field of view (blue-stained part is collagen fibers, and black arrows are collagen fibers), n = 3; (B) Fraction of Masson-stained collagen volume, **p < 0.01 vs. Control; ##p < 0.01 vs. DC. (C, D) Western blot analysis of the protein expression changes of MMP3, MMP8, MMP13, and CollagenIII in the myocardial tissue of the rats in the respective group. n = 3, *p < 0.05 vs. Control; #p < 0.05 vs. DC. (E) RT-qPCR detection of the expression of Col3a1 in the myocardial tissue of the rats. n = 3, **p < 0.01 vs. Control; ##p < 0.01 vs. DC, Take GAPDH as reference. Values are presented as mean ± SD deviation. DC, diabetic cardiomyopathy.
|
SO2 can inhibit cardiomyocyte apoptosis induced by high glucose
In this paper, we observed the effect of high glucose on the apoptosis of H9c2 cardiomyocytes and the protective effect of SO2. Flow cytometry was used to detect the apoptosis rate of cells in each group (Fig. 4A). It was found that the apoptosis of H9c2 cardiomyocytes in the HG group increased by about 4.5 times compared with the control group under high-glucose stimulation. The incidence of early and late apoptotic cells was significantly increased (p < 0.05). After the intervention of exogenous SO2, the apoptosis rate of cardiomyocytes in the HG + SO2 group decreased by about 1/2, and the late apoptotic cells decreased significantly (p < 0.05). At the same time, the apoptosis-associated proteins were detected through Western blot (Fig. 4B–F): Bax/Bcl2, Cleaved Caspase3/Caspase3, Cleaved Caspase9/Caspase9 in the HG group were significantly higher than those in Control group (p < 0.05). After SO2 intervention, the above apoptotic proteins were significantly down-regulated (p < 0.05), and HDX could significantly reverse the anti-apoptotic effect of SO2. The expressions of Bax/Bcl2, Cleaved Caspase3/Caspase3, and Cleaved Caspase9/Caspase9 in cardiomyocytes of the HG + SO2 + HDX group were up-regulated (p < 0.05). The above results suggest that the high-glucose environment can induce the apoptosis of H9c2 cardiomyocytes, while SO2 intervention can reduce the apoptosis of cardiomyocytes induced by high-glucose stimulation.
 | Fig. 4SO2 inhibits high-glucose-induced apoptosis in cardiac myocytes.(A) Flow cytometry to detect the apoptosis rate of H9c2 cardiomyocytes under high glucose environment (33 mM), Q2 + Q4 represents the incidence of apoptosis, n = 3; (B–F) The expression changes of apoptosis-associated proteins Bax, Bcl2, Caspase3, Cleaved-Caspase3, Caspase9, and Cleaved-Caspase9 in myocardial cells of each group were detected through Western blot. n = 3. Values are presented as mean ± SD. SO2, sulfur dioxide; HG, high glucose. *p < 0.05 vs. Control; #p < 0.05 vs. HG; $p < 0.01 vs. HG + SO2.
|
Similar changes were observed in in vivo experiments. By TUNEL staining on the myocardial tissue of diabetic rats (Fig. 5A, B), we found that the brown-stained apoptotic cardiomyocytes in the myocardial tissue of diabetic rats in the DC group increased by about 1.76 times compared with the control group (p < 0.05), after SO2 intervention, TUNEL-positive apoptotic cardiomyocytes in the myocardial tissue were significantly reduced compared with DC group (p < 0.05). Compared with the Control group, the ratio of apoptosis-associated proteins Bax/Bcl2, Cleaved Caspase3/Caspase3, and Cleaved Caspase9/Caspase9 in the DC group increased significantly (p < 0.05), and the mRNA expression levels of Caspase3 and Bax were about 3 times higher than those in the Control group (Fig. 5H, I), while the changes of the above proteins in DC + SO2 group were significantly reversed (p < 0.05).
 | Fig. 5SO2 reduces cardiomyocyte apoptosis in diabetic rats.(A) TUNEL staining of myocardial tissue of the rats in the Control, DC, DC + SO2 and SO2 groups (apoptosis-positive cells in TUNEL staining are in brown, and they are indicated by black arrows), n = 3. Images were acquired at 10 × 40 magnification; (B) Quantitative results of TUNEL-stained positive cells in the myocardial tissue of the rats in the respective group, **p < 0.01 vs. Control. ##p < 0.01 vs. DC; (C–G) Western blot detection of the expressions of Bax, Bcl2, Cleaved Caspase3, Caspase3, Cleaved Caspase9, Caspase9 in the myocardial tissue of the rats in the respective group. n = 3, *p < 0.05 vs. Control; #p < 0.05 vs. DC; (H, I) Detection of mRNA expression levels of Bax and Caspase3 in the myocardial tissue by RT-qPCR, **p < 0.01 vs. Control. ##p < 0.01 vs. DC, GAPDH is taken as the internal parameter. Values are presented as mean ± SD. SO2, sulfur dioxide; DC, diabetic cardiomyopathy.
|
SO2 inhibits apoptosis and fibrosis in cardiomyocytes through upregulation of autophagy
The expression of autophagy-associated proteins in the myocardial tissue of the rats in the respective group was observed through Western blot. As revealed by the results (Fig. 6B, C), the expressions of Atg5, Atg16L1, LC3II/I, and Beclin1 in the myocardial tissue of the DC group were significantly lower than those of the Control group (p < 0.05). While the expression of P62 was significantly up-regulated (p < 0.05), and the above protein changes were reversed (p < 0.05) after SO2 intervention. RT-qPCR results showed that the expression level of P62 in the DC group was about 3.5 times higher than that in the Control group (Fig. 6G), and this change could be down-regulated after SO2 intervention. Moreover, it was found through transmission electron microscopy (Fig. 6A) that compared with the control group, the myocardial tissues of the DC group were characterized by vacuolized autophagosomes, fewer autophagosomes, swollen mitochondria, and disordered arrangement of muscle fibers. Besides, the number of autophagosomes in the myocardial tissue of the rats after SO2 intervention increased, vacuolization was rarely observed, and the arrangement of muscle fibers was relatively neat. The changes in autophagy-associated protein expression in the respective group of H9c2 cardiomyocytes were examined through Western Blot and immunofluorescence in vitro experiments (Fig. 6D–F) and found that autophagy-associated proteins (including LC3II/I, Beclin1, and Atg16L1) were significantly down-regulated (p < 0.05), the fluorescence intensity of Beclin1 was down-regulated in the HG group, and P62 was significantly up-regulated under high-glucose stimulation compared with the Control group (p < 0.05). However, after SO2 intervention, the expression of LC3II/I, Beclin1, and Atg16L1 was significantly up-regulated (p < 0.05), and P62 was significantly down-regulated (p < 0.05) compared with the results of the HG group, while the fluorescence intensity of Beclin1 was significantly attenuated. HDX intervention could reverse the regulation effect of SO2 on the above autophagy-associated proteins (p < 0.05). High glucose was found to lead to the decrease of the autophagy capacity of cardiomyocytes, while SO2 intervention could partially up-regulate or restore autophagy.
 | Fig. 6Sulfur dioxide (SO2) can up-regulate the level of autophagy in the myocardial tissue of type II diabetes rats.(A) The ultrastructure of cardiomyocytes and the changes of autophagosomes in type II diabetes rats were observed under transmission electron microscope. The arrows indicate autophagosomes, and the part indicated by the arrows in the upper figure is enlarged in the lower figure. The TEM figure was observed under ×5,000 at 2 µm bar. (B, C) Western blot analysis of the expression changes of autophagy-associated proteins (including LC3II/I, Beclin1, Atg5, Atg16L1 and P62) in the myocardial tissue of the rats in the respective group. n = 3, *p < 0.05 vs. Control. #p < 0.05 vs. DC. (D, E) Western blot analysis of the expression changes of LC3II/I, Beclin1, Atg16L1 and P62 of H9c2 cardiomyocytes in the Control, HG, HG + SO2, HG + SO2 + HDX and SO2 groups. n = 3; *p < 0.05 vs. Control. #p < 0.05 vs. HG. $p < 0.05 vs. HG + SO2. (F) Immunofluorescence observation of Beclin1 expression in H9c2 cardiomyocytes from each group (magnification; ×10). (G) RT-qPCR detection of P62 mRNA expression in rat myocardial tissue, *p < 0.05 vs. Control; #p < 0.01 vs. DC, take GAPDH as a reference. Values are presented as mean ± SD. DC, diabetic cardiomyopathy; HG, high glucose.
|
SO2 activates autophagy to antagonize cardiomyocyte apoptosis by downregulating PI3K/AKT
In this paper, the changes in PI3K/AKT protein expression in the myocardial tissue of the rats in the respective group were observed through Western blot (Fig. 7A, C). As revealed by the results, compared with the Control group, the expressions of P-PI3K and P-AKT proteins in the DC group were significantly up-regulated (p < 0.05), and the ratios of P-PI3K/PI3K and P-AKT/AKT were also significantly increased. After SO2 intervention, the expressions of P-PI3K and P-AKT proteins in the myocardium of the rats in the DC + SO2 group were down-regulated (p < 0.05), and the ratios of P-PI3K/PI3K and P-AKT/AKT were decreased. There was no significant difference in the expression of P-PI3K and P-AKT between the SO2 control group and the Control group (p > 0.05).
 | Fig. 7Sulfur dioxide (SO2) can reverse the up-regulation of phosphatidylinositol 3-kinase (PI3K)/AKT expression induced by high glucose.(A, C) Western blot detection to observe the expression changes of P-PI3K, PI3K, P-AKT, AKT in the myocardial tissues of the rats in the Control, DC, DC + SO2, and SO2 groups. n=3; *p < 0.05 vs. Control; #p < 0.01 vs. DC. (B, D) Western blot detection was used to observe the protein expression changes of P-PI3K, PI3K, P-AKT, and AKT of H9c2 cardiomyocytes in the Control, HG, HG + SO2, HG + SO2 + 740 Y-P and SO2 group. n = 3; *p < 0.05 vs. Control. #p < 0.05 vs. HG. $p < 0.05 vs. HG + SO2. Values are presented as mean ± SD. DC, diabetic cardiomyopathy; HG, high glucose.
|
The results of the in vitro experiments also suggested that compared with the Control group (Fig. 7B, D), the P-PI3K and P-AKT proteins in the cardiomyocytes of the HG group were up-regulated (p < 0.05), while the ratios of P-PI3K/PI3K and P-AKT/AKT increased. After SO2 intervention, the P-PI3K and P-AKT proteins in the HG + SO2 group were down-regulated (p < 0.05), while the ratios of P-PI3K/PI3K and P-AKT/AKT were all decreased. At the same time, to further study whether SO2 regulates autophagy and apoptosis via the PI3K/AKT pathway, we used 740 Y-P (30 μM), an agonist of PI3K/AKT for intervention. It was found that the levels of LC3II/I, Beclin1, and Atg16L1 of cardiomyocytes in the HG + SO2 + 740 Y-P group were significantly lower than those in the HG + SO2 group (p < 0.05), and the level of P62 increased significantly (p < 0.05). The levels of Cleaved Caspase3/Caspase3, Cleaved Caspase9/Caspase9, and Bax/Bcl2 were significantly higher than those in the HG + SO2 group (p < 0.05), as shown in Fig. 8C–G, which revealed that 740 Y-P, an agonist of PI3K/AKT, could reverses SO2 and down-regulates PI3K/AKT to activate autophagy to antagonize hyperglycemia-induced apoptosis in cardiomyocytes, while SO2 antagonized cardiomyocyte apoptosis by down-regulating PI3K/AKT and activating autophagy.
 | Fig. 8Sulfur dioxide (SO2) antagonizes apoptosis of H9c2 cardiomyocytes by down-regulating phosphatidylinositol 3-kinase (PI3K)/AKT and activating autophagy.(A, B) Western blot detection was used to observe the expression changes of autophagy-associated proteins (including LC3II/I, Beclin1, Atg16L1 and P62 of H9c2 cardiomyocytes) in the Control group, the HG group, the HG + SO2 group, the HG + SO2 + 740 Y-P group and the SO2 group. (C–G) Western blot detection was used to observe the expression changes of apoptosis-associated proteins Bax, Bcl2, Caspase3, Cleaved-Caspase3, Caspase9, and Cleaved-Caspase9 of H9c2 cardiomyocytes in the Control, HG, HG + SO2, HG + SO2 + 740 Y-P, and SO2 groups. n = 3. Values are presented as mean ± SD. DC, diabetic cardiomyopathy; HG, high glucose. *p < 0.05 vs. Control. #p < 0.05 vs. HG. $p < 0.05 vs. HG + SO2.
|
Go to :
DISCUSSION
Type II diabetes is a metabolic disease characterized by hyperglycemia and insulin resistance. It has been recognized as a vital public health problem worldwide and has posed a serious hazard to people’s health and quality of life [32-34]. Cardiomyopathy damage caused by diabetes has been found as one of the main causes of death in type II diabetes patients. Diabetic cardiomyopathy refers to a major cardiovascular disease that is characterized by myocardial abnormalities due to diabetes. Hyperglycemia and insulin resistance, two independent risk factors for DC, can cause abnormal myocardial metabolism and dysfunction and gradually develop into heart failure with the progression of the disease [35,36]. Existing studies have suggested that tissue inflammation, oxidative stress, and dysregulation of autophagy induced by high-glucose stimulation can promote dysfunction of cardiomyocytes and fibrosis of the myocardial interstitium, thus leading to diabetic cardiomyopathy [37,38]. Myocardial fibrosis is a vital pathological change leading to diabetic cardiomyopathy, which is primarily manifested as myocardial pathological changes characterized by excessive deposition of myocardial extracellular matrix and myocardial structural disorder; it can develop into heart failure [39-41]. In this experiment, a model of rats with type II diabetes was established by a diet high in fat and sugar and through the intraperitoneal injection of STZ. The results of Masson staining showed that the DC group formed more collagen fibers than the normal group. Besides, the ultrasound results indicated that the cardiac function was lower than that of the normal group, thus revealing that myocardial fibrosis could cause obvious myocardial fibrosis and cardiac dysfunction in diabetic rats.
Apoptosis has been found as a key link between myocardial fibrosis and myocardial remodeling [42]. Since cardiomyocytes are permanent cells, it is difficult to repair and rebuild through proliferation and differentiation after myocardial injury, and effectively antagonizing cardiomyocyte apoptosis will help to improve myocardial fibrosis and inhibit myocardial remodeling [43]. The experimental result showed that the number of positive cells in the myocardium of diabetic rats after TUNEL staining increased significantly, and the pro-apoptotic protein in the myocardial tissue was significantly up-regulated compared with the Control group. The result of the in vitro experiments suggested that high-glucose stimulation could induce the apoptosis of H9c2 cardiomyocytes. Furthermore, the expression of pro-apoptotic proteins (Cleaved Caspase3, Cleaved Caspase9, and Bax) of H9c2 cardiomyocytes in the HG group was significantly up-regulated as compared with the Control group. As revealed by the above results, the high-glucose environment could promote cardiomyocyte apoptosis, and apoptosis might be a vital factor leading to myocardial fibrosis in diabetes rats, consistent with the study by Zou and Xie [44].
Autophagy means that cells engulf their own damaged organelles or proteins and fuse with lysosomes to meet the renewal and metabolic needs of the organelles of cells. Moderate autophagy is an important self-protection mechanism of damaged cells under stress [45], which helps maintain cellular physiological homeostasis. Insufficient autophagy may lead to the dysfunction of cardiomyocyte self-repair, which in turn leads to cardiomyocyte injury and even induces apoptosis [46]. Previous studies have suggested that autophagy is one of the important causes of diabetic cardiomyopathy. Autophagy-dependent cell death leads to the loss of cardiomyocytes and initiates the repair program of fibrosis, which is an important factor in the occurrence of diabetic cardiomyopathy [47]. However, recent studies have found that moderate autophagy can antagonize cellular inflammation, mitochondrial stress, oxidative stress, etc., thereby inhibiting cardiomyocyte apoptosis [48]. Dewanjee et al. [15] highlighted that autophagy is significantly correlated with diabetes-induced cardiomyopathy and may be a vital intervention target for diabetic cardiomyopathy. Previous studies have found that the disorder or deficiency of autophagy can induce cardiomyocyte apoptosis in diabetic rats/mice [17,49,50]. Our previous study also found that the level of autophagy in cardiomyocytes of diabetic rats was down-regulated, and up-regulation of autophagy could antagonize high glucose-induced cardiomyocyte senescence [16]. In this study, we observed by transmission electron microscopy and immunoblotting that: autophagosomes, LC3II/I, Beclin1, and other autophagy-associated proteins in cardiomyocytes of diabetic rats were significantly down-regulated compared with those of the Control group. Through in vitro experiments, we also found that the level of autophagy in cardiomyocytes under high glucose stimulation was significantly down-regulated, which is consistent with other scholars and previous related studies. The down-regulation of autophagy can cause the dysfunction of cellular self-repair and induce cardiomyocyte apoptosis, which in turn leads to myocardial fibrosis.
The PI3K/Akt pathway has been found as a main regulatory pathway of autophagy. Existing studies have suggested that down-regulating the expression of the PI3K/AKT pathway can reduce diabetes-induced cardiac dysfunction and ameliorate myocardial fibrosis [24]. Its mechanism may be correlated with the inhibition of collagen synthesis in cardiac fibroblasts and cardiomyocyte hypertrophy [51]. Over-activation of the PI3K/AKT pathway can induce apoptosis, while down-regulation of PI3K/AKT can inhibit cardiomyocyte apoptosis and improve myocardial fibrosis [23], but the specific mechanism is still unclear. It has been found that inhibiting the excessive activation of PI3K/AKT can restore or upregulate the impaired autophagic flux and antagonize cardiomyocyte apoptosis [20]. In vitro experiments have also found that high-glucose stimulation can promote the phosphorylation of AKT and mTOR in H9c2 cardiomyocytes [13,52], inhibit autophagy, and promote oxidative stress and apoptosis. In the present study, we found that high-glucose stimulation induced apoptosis of H9c2 cardiomyocytes, accompanied by autophagy impairment. Western-blot detection also observed that the levels of P-PI3K and P-AKT in the HG group were significantly higher than those in the Control group, which was consistent with the previous research results of other scholars. It was also found in in vivo experiments that the expression levels of P-PI3K and P-AKT in the fibrotic myocardial tissue of diabetic rats were significantly increased, while the autophagy flux was decreased, and apoptosis-related proteins were significantly increased. It was suggested that the phosphorylation and excessive activation of PI3K/AKT might inhibit autophagy and induce apoptosis, thereby leading to diabetic myocardial fibrosis.
SO2 was considered a harmful gas molecule in the past. With further research on endogenous SO2, the physiological protective effect of SO2 in the body has been gradually revealed. SO2 has been considered the fourth endogenous gas signal molecule newly discovered after NO, CO, and H2S. It exhibits the functions of vasodilator, anti-oxidation, anti-inflammatory, inhibition of apoptosis, and smooth muscle proliferation while having an important protective effect on the cardiovascular system at a proper concentration [30,53-55]. However, the correlation between SO2 and myocardial fibrosis and remodeling remains unclear, and the internal regulatory mechanism is unknown [56]. The endogenous enzyme AAT of SO2 is widely expressed in tissues and organs, and it has the highest expression level in the myocardium. As an important endogenous regulator of cardiovascular homeostasis, exogenous SO2 donors can upregulate the expression of endogenous SO2-producing enzyme AAT1, enhancing the synthesis of endogenous sulfur dioxide and inhibiting cardiomyocyte apoptosis, reducing inflammatory damage mediated by the NLRP3 inflammasome body and improve cardiac function [29]. We have previously found that exogenous SO2 donors can upregulate the expression of GOT1/AAT1 increasing the synthesis of endogenous sulfur dioxide and improving myocardial fibrosis in rats with type 1 diabetes, and is related to the inhibition of cardiomyocyte apoptosis [26], but the internal regulatory mechanism is not very clear. In addition, some scholars have found that SO2 can also inhibit endoplasmic reticulum stress and improve myocardial fibrosis [57], while it can inhibit the proliferation and migration of fibroblasts [27]. Therefore, some scholars regard endogenous SO2 as a potential and promising anti-fibrotic treatment strategy, but the specific mechanism of action is unclear and needs to be further explored [25]. In this study, it was found that endogenous SO2-producing enzyme AAT1/2 in myocardial tissue of diabetic rats and H9c2 cardiomyocytes stimulated by high glucose decreased significantly, suggesting that the production of endogenous SO2 in cardiomyocytes of diabetic rats was insufficient. At the same time, it was found in the study that administration of exogenous SO2 donor can improve the deposition of collagen fibers in the myocardial interstitium of diabetic rats, while the level of damaged autophagy is significantly up-regulated, and the apoptosis of cardiomyocytes is significantly antagonized. The study also found that the above changes may be related to the excessive activation of the PI3K/AKT pathway antagonized by SO2. To investigate whether SO2 regulates autophagy and inhibits cardiomyocyte apoptosis via the PI3K/AKT pathway, PI3K/AKT activator 740 Y-P was added, while cardiomyocytes were intervened in the high-glucose medium with SO2. The results indicated that 740 Y-P significantly increased the phosphorylation level of PI3K/AKT in H9c2 cardiomyocytes, and the autophagy level of cardiomyocytes in the HG + SO2 + 740 Y-P group decreased significantly, while the proportion of apoptotic cells and apoptosis-associated proteins increased significantly. The above results suggested that 740 Y-P, an activator of PI3K/AKT, could reverse the effect of SO2 on inhibiting PI3K/Akt to activate autophagy and antagonize apoptosis, and SO2 might inhibit cardiomyocyte apoptosis by antagonizing PI3K/AKT phosphorylation and increasing autophagic flux, which would ameliorate diabetic myocardial fibrosis.
In brief, this paper found that SO2, a novel gas signaling molecule, could inhibit cardiomyocyte apoptosis by activating autophagy, thus improving myocardial fibrosis and myocardial remodeling in diabetic rats. Moreover, the mechanism might be correlated with the excessive activation of the PI3K/AKT pathway that it antagonized. This study shows that endogenous SO2 might play an important protective role in the occurrence and development of myocardial fibrosis in type 2 diabetes. Likewise, the down-regulation of endogenous SO2 might play a certain role in the mechanism of myocardial remodeling, and the PI3K/AKT-mediated autophagy and its secondary myocardial apoptosis might be an essential link. This paper is expected to discover novel intervention targets for myocardial remodeling and heart failure, and more specific mechanisms (e.g., the mechanism by which SO2 regulates PI3K/AKT phosphorylation) should be studied in depth.
Go to :
 XML Download
XML Download