

 PDF
PDF Citation
Citation Print
Print


INTRODUCTION
METHODS
Materials
Cell culture
Cell viability
Nuclear extraction
Assessment of renin activity and content by RIA and ELISA
Western blot analysis
Confocal immunofluorescence microscopy
Imaging of cytoplasmic renin and its secretion
Statistical analysis
RESULTS
Cell growth and N-cad expression
 | Fig. 1Growth curve and cadherin expression before (lane 1) and after confluence (lane 2).(A) Growth curve. Cells were plated (3 × 103 cells/well) onto 24-well plates (day 0), cultured for 10 days and counted on a hemocytometer every 2–3 days (n = 3). (B) Phase contrast microscopy of ~70% and 100% confluent cells (×200). Scale bar: 60 μm. (C) Expression of cadherins. Protein (40 μg) obtained from cell lysates of cells grown to ~70% confluence (lane 1) or from cells on day 2 postconfluence (lane 2) were resolved by 7% acrylamide SDS-PAGE, followed by Western blotting with antibodies against epithelial cadherin (E-cad), placental cadherin (P-Cad), or neural cadherin (N-Cad). The numbers on the left indicate the molecular masses of standard proteins in kilo Daltons (kDa). The density of the bands was compared by unpaired Student’s tests (n = 3 for each group). β-actin was used as the protein loading control for each gel. NS, no significant difference. (D) N-cad expression at ~70% confluence (upper panel) and 100% confluence (lower panel). Cell nuclei were stained with propidium iodide (DAPI). Scale bar: 10 μm.
|
Effect of confluence on active and inactive renin secretion
Table 1
| ng ANG I/well/h | Inactive/total (%) | |||
|---|---|---|---|---|
| Total | Active | Inactive | ||
| DMEM + 10% FBS | 1,486 ± 209 | 36.8 ± 4.5 | 1,449 ± 209 | 97.5 ± 0.4 |
| DMEM + 10% FBS + ML-7 | 1,502 ± 209 | 26.9 ± 2.9 | 1,475 ± 220 | 98.2 ± 0.4 |
Values represent means ± SD from 6 wells. Preconfluent (70%–80% confluence) As4.1cells were incubated in DMEM + 10% FBS in the absence and presence of ML-7 (6 × 10−5 M) for 1 h each at 37°C in a humidified atmosphere of 5% CO2. Collected media were centrifuged at 1,000 ×g for 10 min at 4°C to remove detached cells. Renin activity was determined with (total) and without (active) trypsin (1 mg/ml) after 30 min on ice. Inactive renin was calculated by subtracting active renin from the total. ANG I, angiotensin I; DMEM, Dulbecco's modified eagle medium; FBS, fetal bovine serum. No significant difference before vs. after ML-7 by paired Student’s t-test.
![]()
Table 2
| Post-confluent day (ng ANG I/well/day) | ||||
|---|---|---|---|---|
| Day 1 | Day 2 | Day 3 | Day 4 | |
| DMEM | 2.68 ± 1.03 | 10.00 ± 3.53 | 6.94 ± 2.82 | 3.10 ± 0.47 |
| DMEM + 10% FBS | 5.33 ± 1.74* | 9.00 ± 5.12 | 13.40 ± 1.65 | 8.29 ± 4.04* |
Values are means ± SD from 6 wells. When cells grown to 100% confluent, incubation media were switched with fresh media of DMEM or DMEM + 10% FBS every day, and incubated for 1 h. Secreted renin activity was determined as described in Table 1. ANG I, angiotensin I; DMEM, Dulbecco's modified eagle medium; FBS, fetal bovine serum. *p < 0.005 DMEM vs. DMEM + 10% FBS.
![]()
Table 3
| Pre-confluence | Postconfluence | |||
|---|---|---|---|---|
| DMEM + 10% FBS | DMEM + 10% FBS | IGF-I | IGF-I + Antibody | |
| Renin secretion (ng/well/h) | 10.5 ± 3.47 | 31.6 ± 1.25* | 73.9 ± 5.72** | 20.1 ± 1.70*** |
Values represent means ± SD from five wells. As4.1 cells were cultured in DMEM + 10% FBS to pre-confluence (70%–80% confluence) or day 2 postconfluence (postconfluence). Alternatively, cells were cultured in DMEM + IGF-I (2.6 × 10−10 M) or DMEM + IGF-I (2.6 × 10−10 M) + antibodies against IGF-I (10 μg/ml) to day 2 postconfluence (postconfluence). Culture media were replaced with fresh media and incubated for 1 h to measure the rate of renin secretion by ELISA. DMEM, Dulbecco's modified eagle medium; FBS, fetal bovine serum. p-values were calculated using the unpaired Student’s t-test. *p < 0.001 DMEM + 10% FBS at Pre-confluence vs. DMEM + 10% FBS at Postconfluence; **p < 0.001 DMEM + 10% FBS at Postconfluence vs. DMEM + IGF-I at Postconfluence; ***p = 0.001 DMEM + IGF-I at Postconfluence vs. DMEM + IGF-I + Antibody at Postconfluence.
![]()
Table 4
| Pre-confluence | Postconfluence | |||
|---|---|---|---|---|
| + FBS (1) | – FBS (2) | + FBS (3) | + FBS + IGF-I antibody (4) | |
| Active renin (ng/well/h) | 7.2 ± 0.52 | 10.9 ± 0.31* | 17.9 ± 0.47** | 10.3 ± 0.34***,**** |
| Inactive renin (ng/well/h) | 366 ± 112 | 245 ± 36.7† | 51.2 ± 16.0†† | 242 ± 38†††, †††† |
Values represent means ± SD from 6 wells. Cells were cultured to 70%–80% confluence in DMEM + 10% FBS (Pre-confluence) or to 100% confluence. Culture medium was replaced with DMEM without FBS (– FBS), DMEM + 10 % FBS (+ FBS), or DMEM + 10% FBS containing 10 ng/ml IGF-I antibody and cultured for 2 more days (Postconfluence). Cells were incubated in fresh media under each culture condition for 1 h to determine the rate of renin secretion. Levels of active and inactive renin were determined with the use of mouse renin ELISA kit from RayBiotech® and mouse prorenin kit from Molecular Innovations®, respectively. DMEM, Dulbecco’s modified eagle medium; FBS, fetal bovine serum; IGF-I, insulin-like growth factor-I. p-values were calculated by ANOVA. *p < 0.001 (1) vs. (2); **p < 0.001 (2) vs. (3); ***p < 0.001 (3) vs. (4); ****p < 0.001 (1) vs. (4); †p = 0.006 (1) vs. (2); ††p < 0.001 (2) vs. (3); †††p < 0.001 (3) vs. (4); ††††p = 0.005 (1) vs. (4). No significant difference between (2) and (4) in active and inactive renin secretion.
![]()
Identification of FBS components that modulates renin secretion
Relationship between active renin and inactive prorenin secretion
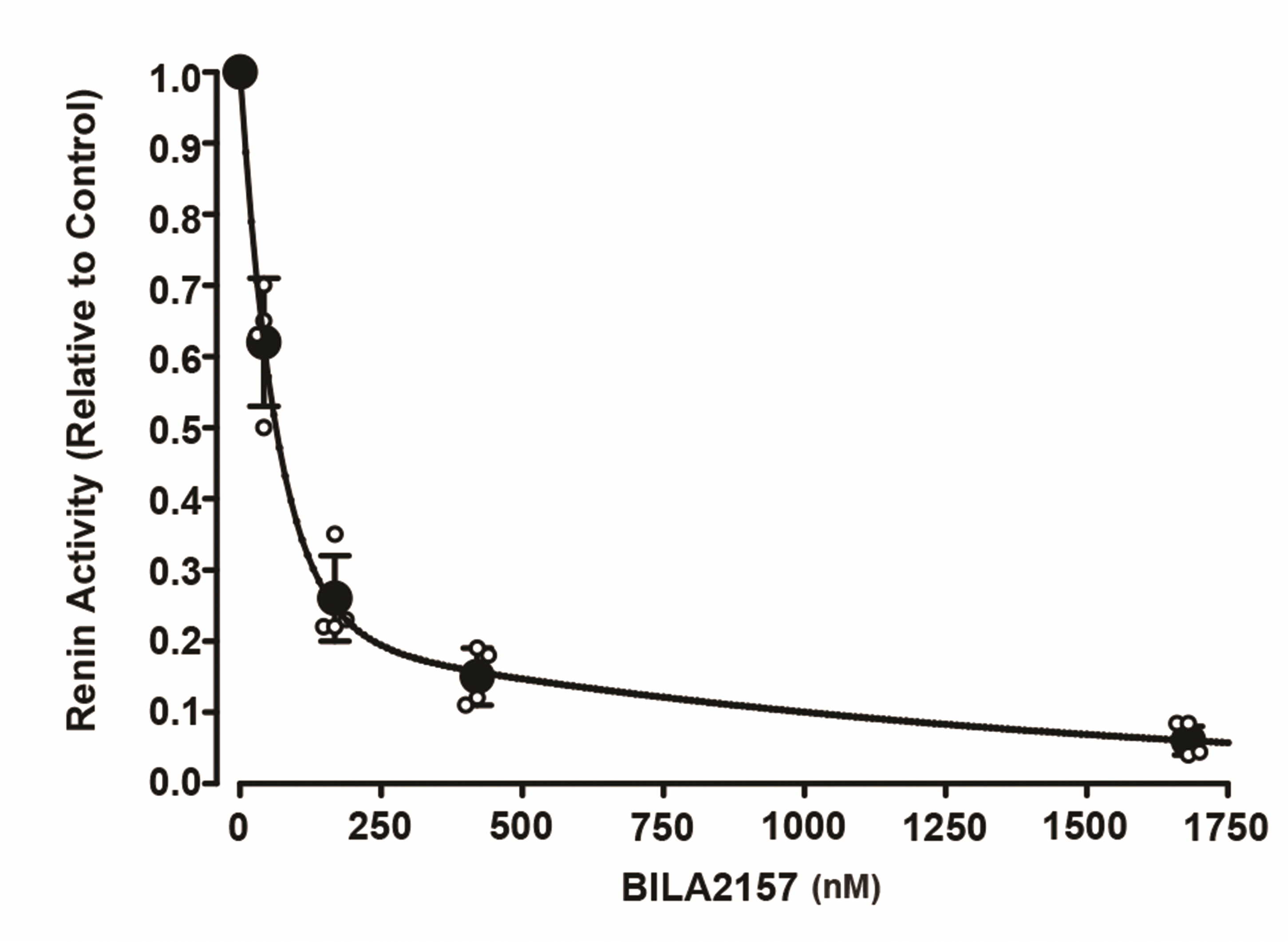 | Fig. 2BILA 2157 BS inhibits renin activity.Incubation media from postconfluent cells with a renin activity of 268.3 ± 51 ng ANG I/ml/h (relative value 1.0) was incubated with varying concentrations of BILA 2157 BS for 1 h. Active renin activity was determined by radioimmunoassay for ANG I. Each data point represents the mean ± SD of three samples. ANG I, angiotensin I.
|
 | Fig. 3Effects of Ca2+, calmidazolium, ML-7, isoproterenol and forskolin on active renin secretion in postconfluent As4.1 cells.Cells on day 2 postconfluence were incubated in Ca2+-free DMEM containing 1 mM EGTA (–Ca2+ DMEM) (A, left panel) and then in 2 mM Ca2+-containing DMEM and 10% FBS (+Ca2+ DMEM) successively for 1 h each or in the reverse order (A, right panel, p < 0.001, n = 6). In the next series of experiments, cells were incubated in +Ca2+ DMEM for 1 h (control) and then another 1 h in +Ca2+ DMEM + 10 % FBS containing either calmidazolium (3 × 10−5 M) (B, n = 6), ML-7 (6 × 10−5M) (C, n = 6), isoproterenol (10−8–10−5 M) (D, n = 6) or forskolin (3 × 10−5 M) plus IBMX (10−4 M) (E, n = 6). The rate of active renin secretion was determined by radioimmunoassay for ANG I in (A) and (B) or by ELISA in (C), (D), and (E). ANG I, angiotensin I; DMEM, Dulbecco’s modified eagle medium; FBS, fetal bovine serum. Samples within groups were compared using paired Student’s t-tests and between groups using unpaired Student’s t-tests (A–C, E) or ANOVA (D), *p < 0.001 vs. control, except between samples at the two highest concentrations, which were compared by unpaired Student’s t-tests. †p < 0.001.
|
Table 5
| Control | ML-7 | Forskolin | Calyculin A | |
|---|---|---|---|---|
| Prorenin (ng/well/h) | 81.5 ± 7.6 | 21.4 ± 1.1*,** | 36.3 ± 9.7*,** | 155.2 ± 20.7* |
Values represent means ± SD from 6 wells. On day 2 postconfluence, cells were incubated in DMEM + 10% FBS alone (Control) or containing ML-7 (6 × 10−5 M), forskolin (3 × 10−5 M) + IBMX (10−4 M), or calyculin A (2 × 10−7 M) for 1 h. Levels of secreted prorenin were determined with the use of mouse prorenin/renin total antigen ELISA kit from Molecular Innovations. DMEM, Dulbecco's modified eagle medium; FBS, fetal bovine serum. p-values were calculated by ANOVA. *p < 0.001 vs. Control; **p < 0.001 vs. Calyculin A. No significant difference between ML-7 and Forskolin.
![]()
Identification of an ANG I-generating enzyme in As4.1 cell incubation medium
Effects of known stimuli on active renin secretion
Table 6
| Agent | Action | n | Effect on active renin secretion (E/C) |
|---|---|---|---|
| Ionomycin (10−5 M) | Ca2+ ionophore | 6 | 0.57 ± 0.12* |
| ANG II (10−8 M) | Phospholipase activator | 7 | 0.54 ± 0.16* |
| U73122 (3 × 10−5 M) | PLC inhibitor | 5 | 4.23 ± 1.63* |
| U73343 (3 × 10−5 M) | Inactive analogue of U 73122 | 5 | 1.19 ± 0.38 |
| Ophiobolin A (3 × 10−5 M) | CaM inhibitor | 6 | 3.76 ± 1.22* |
| K252a (10−5 M) | Broad spectrum protein kinase inhibitor | 6 | 2.33 ± 0.93* |
| Butanedione monoxime (25 mM) | MLCK inhibitor | 5 | 4.35 ± 0.85* |
| GF-109203X (3 × 10−6 M) | PKC inhibitor | 6 | 0.97 ± 0.15 |
| Y-27632 (3 × 10−5 M) | Rho kinase inhibitor | 8 | 1.34 ± 0.11* |
| Calyculin A (2 × 10−7 M) | Protein phosphatase 1 & 2A | 6 | 0.77 ± 0.12* |
| LY294002 (5 × 10−5 M) | Phosphatidylinositol-3 kinase inhibitor | 5 | 0.61 ± 0.13* |
| Wortmanin (10−6 M) | Phosphatidylinositol-3 kinase inhibitor | 5 | 0.54 ± 0.09* |
| SB216763 (2 × 10−5 M) | GSK-3β inhibitor | 6 | 1.89 ± 0.17* |
| Blebbistatin (10−6 M) | Myosin ATPase inhibitor | 8 | 1.21 ± 0.37 |
Values represent means ± SD. Cells were grown in DMEM + 10% FBS to 100% confluence. On day 2 postconfluence, cells were incubated in 1 ml fresh DMEM + 10% FBS for 1 h (C) followed by incubation in medium containing one of the above agents (E). Concentrations in parentheses are the concentrations tested. (E/C) values significantly greater than 1.0 or less than 1.0 indicate stimulation or inhibition of renin secretion, respectively. DMEM, Dulbecco's modified eagle medium; FBS, fetal bovine serum. *p < 0.05.
![]()
Postconfluent phenotypic switching of As4.1 cells in the presence and absence of FBS
 | Fig. 4Expression of N-cadherin, MRTF-A, sm MHC, nm MHC and PP1β changes to As4.1 cells before and after confluence.Cell lysates from cells grown to 70%–80% confluence in the presence of 10% FBS (A, lane 1), cells at day 2 postconfluence in the continuous presence of 10% FBS (A, lane 2) or in the absence of 10% FBS for two days after achieving 100% confluence (A, lane 3). After the aspiration of the incubation media, cells were lysed and centrifuged. Supernatant protein (60 μg) was resolved by SDS-PAGE on 7% or 15% acrylamide slab gels followed by Western blotting for N-cad, MRTF-A, sm MHC, nm MHC, and PP1β and then analyzed by densitometry. In the case of MRTF-A, a nuclear fraction was prepared, and 100 μg was resolved (B, upper panel). p-values were obtained by ANOVA. To assess nuclear localization of MRTF-A, cells were plated onto coverslips and fixed and permeabilized as described in the Methods section. Cells were then immunostained with primary MRTF-A antibody (1:100) and then with FITC-labelled goat anti-mouse secondary antibody to MRTF-A (1:1,000). Nuclei were stained with propidium iodide (DAPI) (B, lower panel). The density of preconfluent cells was arbitrarily set to 1. Fluorescence was analyzed by an image analyzer. Lane 1, preconfluent cells; lane 2, postconfluent cells in the presence of 10% FBS. p-values were obtained by unpaired Student’s t-tests (n = 4). Scale bar: 10 μM. (C) Postconfluent cells were incubated in DMEM + 10% FBS with or without CCG-1423 (5 × 10−6 M), an inhibitor of nuclear translocation of MRTF-A, for 1 h and then with ML-7 (6 × 10−5 M) for another 1 h. Secreted renin was measured by ELISA (C, n = 5). MRTF-A, myocardin related transcription factor-A; sm MHC, smooth muscle myosin heavy chain; nm MHC, nonmuscle myosin heavy chain; PP1β, protein phosphatase 1β; FBS, fetal bovine serum. NS, no significant difference. p-values within groups were obtained by paired Student’s t-tests, and those between groups were obtained by ANOVA.
|
 | Fig. 5Effects of MRTF-A knockout on phenotypic changes.Cells at ~70% confluence were transfected with control plasmids (A, lane 1) or the MRTF-A gene was knocked out using MRTF-A HDR CRISPR-associated 9 (Cas9) (A, lane 2) for three days. On day 2 postconfluence, the expression of MRTF-A, N-cad, sm MHC, nm MHC, and PP1β in the supernatant fraction (80 μg) was determined as described in the legend of . p-values were obtained by unpaired Student’s t-tests. (B) Control and knockout cells were incubated before and after stimulation with ML-7 (6 × 10−5 M) for 1 h each, and secreted active renin was measured by ELISA (B, n = 4). MRTF-A, myocardin related transcription factor-A; HDR, homology directed repair; CRISPR, clustered regularly interspaced short palindromic repeats; sm MHC, smooth muscle myosin heavy chain; nm MHC, nonmuscle myosin heavy chain; PP1β, protein phosphatase 1β. NS, no significant difference. p-values were obtained either by paired Student’s t-tests (within groups) or by ANOVA (between groups).
|
 | Fig. 6Effects of SRF knockout on phenotypic changes.Cells were transfected with control plasmids (A, lane 1) or SRF HDR CRISPR/Cas9 plasmids (A, lane 2) as described in the legend of . Then, the expression of each protein was determined in the supernatant fraction (80 μg). Both control and knockout cells were incubated with ML-7 (6 × 10−5 M) before and after stimulation for 1 h each, and secreted active renin was measured by ELISA (B, n = 5). SRF, serum response factor; HDR, homology directed repair; CRISPR, clustered regularly interspaced short palindromic repeats. p-values were obtained either by paired Student’s t-tests (within groups) or by ANOVA (between groups).
|
 | Fig. 7Effects of N-cad expression on pMLC20 and PP1β.Cells were cultured to 100% confluence and maintained for two more days in the presence of 10% FBS (lane 1) or in the presence of both FBS and IGF-1 antibody (10 μg/ml) (lane 2). Proteins in the supernatant (40 μg) were resolved by Western blotting as described above (n = 4). pMLC20, phosphorylated 20 kDa myosin light chain; PP1β, protein phosphatase 1β; FBS, fetal bovine serum; IGF-I, insulin-like growth factor-I. p-values were obtained by unpaired Student’s t-tests.
|
 | Fig. 8Effects of MYLK knockdown on phenotypic changes.Cells were transfected with control or siRNA of MYLK plasmids as described in the legend of . Expression of MYLK and the cytosolic pMLC20 level (A) were determined using supernatant protein (60 μg). p-values were obtained by unpaired Student’s t-tests (n = 3). Active renin secretion from control and knockdown cells was determined by incubating cells in DMEM + 10% FBS for 1 h (B, n =6). pMLC20, phosphorylated 20 kDa myosin light chain; DMEM, Dulbecco’s modified eagle medium; FBS, fetal bovine serum. p-values by unpaired Student’s t-tests.
|
 | Fig. 9Effects of PP1β knockdown on phenotypic changes.Cells were transfected with control (lane 1) or siRNA of PP1β plasmids as described in the legend for . The expression of PP1β (A, upper panel) and cytosolic pMLC20 level (A, lower panel) were determined using supernatant protein (60 μg). p-values were obtained by unpaired Student’s t-tests (n = 4). Active renin secretion was determined before and after stimulation by ML-7 (6 × 10−5 M) for 1 h each, and secreted renin was measured by ELISA (B). PP1β, protein phosphatase 1β; pMLC20, phosphorylated 20 kDa myosin light chain. p-values were obtained either by paired Student’s t-tests (within groups) or by ANOVA (between groups) (n = 6).
|
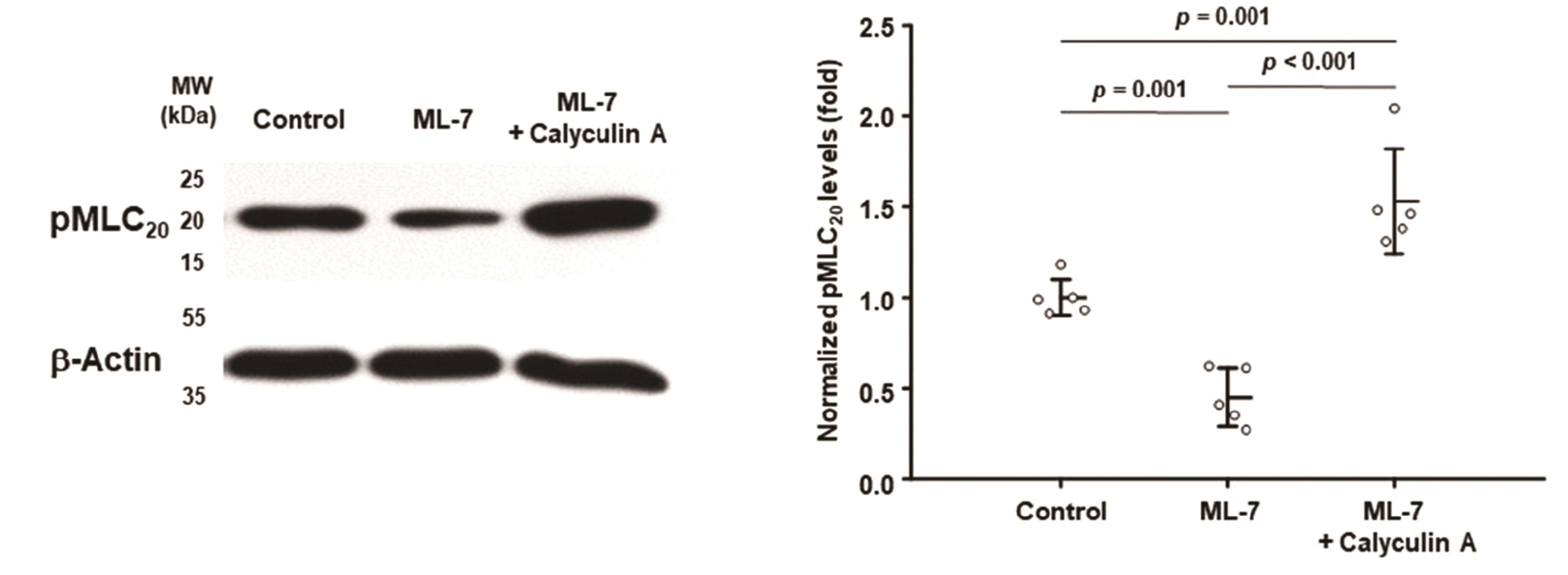 | Fig. 10Effects of ML-7 and calyculin A on the phosphorylation of MLC20.On day 2, postconfluent cells were incubated in DMEM + 10% FBS (control) in the presence of ML-7 (6 × 10−5 M; ML-7) and in the presence of both calyculin A (2 × 10−7 M) and ML-7 (6 × 10−5 M; ML-7 + Caly) for 1 h. The level of pMLC20 was determined using supernatant protein (80 μg). MLC20, 20 kDa myosin light chain; DMEM, Dulbecco’s modified eagle medium; FBS, fetal bovine serum. p-values were obtained by ANOVA (n = 5).
|
Visualization and imaging of renin secretory granules and secretion
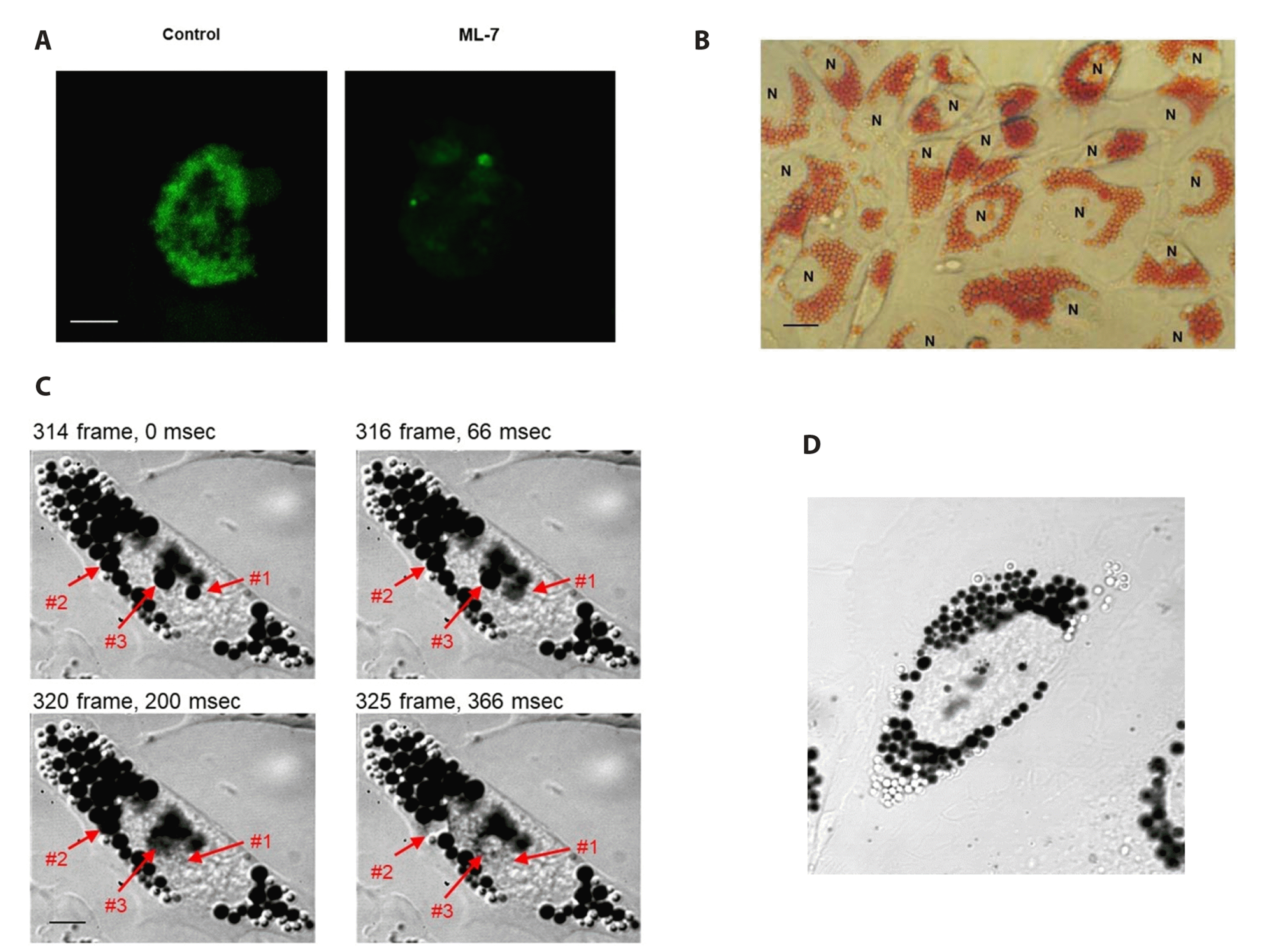 | Fig. 11Imaging of exocytotic discharge of renin and neutral red by ML-7.Cells on postconfluent day 2 were plated on cover slips were permeabilized and stained with antibody to renin. Many cytoplasmic stained granules are seen (A, control). When cells were pretreated with ML-7 (6 × 10−5 M) for 2 min and stained with renin antibody, no granules were observed (ML-7). Scale bar: 10 μm. Cells were incubated with 100 μM neutral red for 3 h, washed with fresh DMEM, and photographed with a light microscope. Numerous neutral red stained pink granules are seen (B). N denotes nucleus. Scale bar: 3.33 μm. (C, D) When neutral red-loaded granules were viewed at the isosbestic absorbent light at 470 nm, the granules observed were black. White granules are likely discharged granules prior to imaging. When cells were perfused with ML-7 (6 × 10−5 M), #1, #2, and #3 neutral red-loaded granules were discharged over 200 msec. (D) The time-lapsed movie shows the discharge of neutral red-loaded secretory granules (Supplementary Video). DMEM, Dulbecco’s modified eagle medium.
|
DISCUSSION
Induction of the regulated renin secretory phenotype
N-cad expression at cell-cell contacts triggers phenotype changes
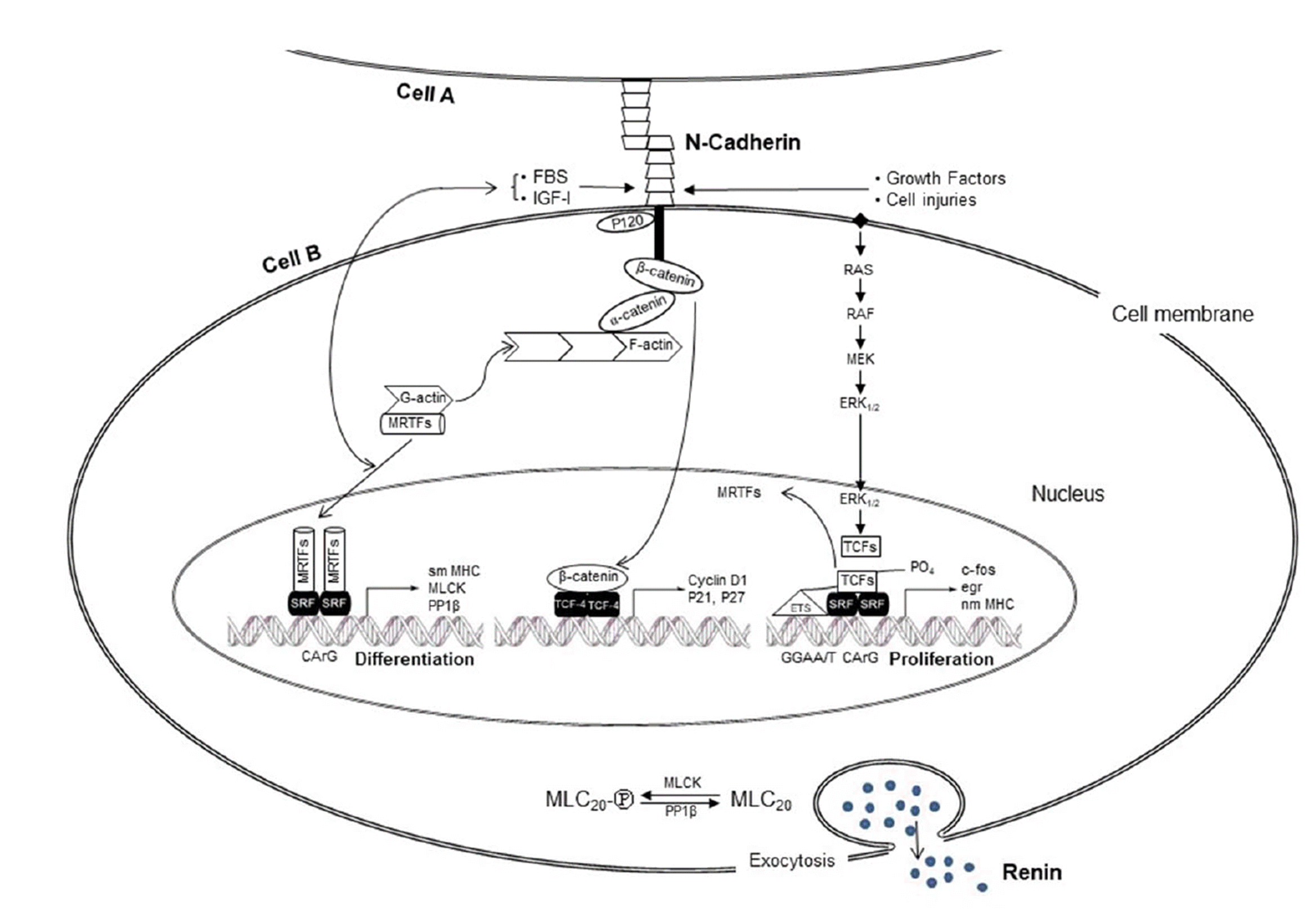 | Fig. 12Schematic summary of the transcriptional cascades regulating expression of proteins associated with renin secretory phenotypes in As4.1 cells.N-cad expression at the plasma membrane upon cell-cell contact triggers MRTF-A-SRF-CArG box, which activates transcription of smooth muscle-specific genes encoding contractile proteins in association with the regulatory phenotype of active renin secretion (left side). Without N-cad expression, β-catenin (signaling cascade in the middle) and growth factors (signaling cascade of right side) trigger a signaling cascade of growth factors of cell proliferation. MRTF-A, myocardin related transcription factor-A; SRF, serum response factor; MLC20, 20 kDa myosin light chain; PP1β, protein phosphatase 1β; FBS, fetal bovine serum; IGF-I, insulin-like growth factor-I; sm MHC, smooth muscle myosin heavy chain; nm MHC, nonmuscle myosin heavy chain.
|
Correlation of expression of specific proteins and induction of regulated renin secretory phenotypes
SUPPLEMENTARY MATERIALS
spaceplay / pause
qunload | stop
ffullscreen
shift + ←→slower / faster
↑↓volume
mmute
←→seek
. seek to previous
12… 6 seek to 10%, 20% … 60%

 XML Download
XML Download