

 PDF
PDF Citation
Citation Print
Print



INTRODUCTION
Polyarteritis nodosa (PAN) is defined as a necrotizing inflammation of small- to medium-sized arteries without glomerulonephritis or vasculitis in the arterioles, capillaries, or venules, which is also not associated with antineutrophil cytoplasmic antibodies [1]. Systemic PAN is characterized by specific signs and symptoms that involve various organs including the skin, muscle, joints, heart, kidneys, bowels, and brain, with constitutional symptoms, such as fever and malaise. The diagnosis of childhood-onset PAN (cPAN) is occasionally delayed because of its nonspecific and varying manifestations [2,3].
Deficiency of adenosine deaminase 2 (DADA2) is a rare autoinflammatory disease caused by biallelic loss-of-function of the ADA2 gene which was first described in two different cohorts for a monogenic vasculitis syndrome similar to PAN in 2014 [2,4]. The main phenotype of DADA2 is vasculitis involving small-to medium-sized arteries, such as in systemic PAN. However, it is accompanied by early-onset strokes, immunological and hematological manifestations, such as hypogammaglobulinemia, lymphopenia, pure red cell aplasia, neutropenia, and thrombocytopenia; and lymphoproliferation, including hepatosplenomegaly [5-8].
Herein, we describe a case of DADA2, that is the first case in Korea, and caused by a compound heterozygous pathogenic variant including splicing variant in the ADA2 gene, in a patient who was previously diagnosed with cPAN.
CASE REPORT
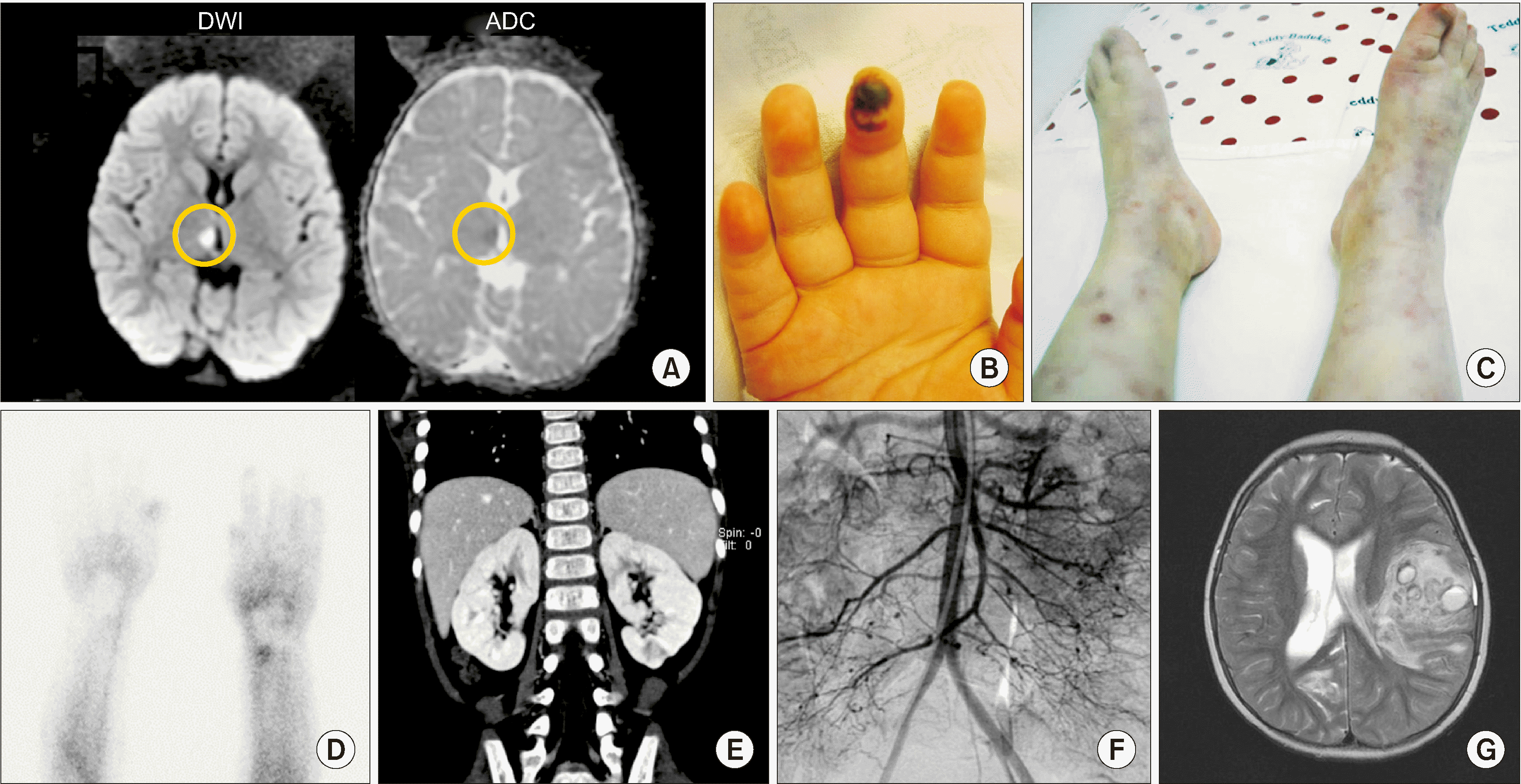
A 35-month-old female patient was referred to Seoul National University Children’s Hospital (SNUCH) due to ptosis on the left side, headache, and fever. At 34 months of age, she had truncal ataxia, hoarseness, along with left side ptosis and facial palsy. Magnetic resonance imaging of the brain showed a thalamic stroke, but there was no evidence of sepsis and infection in the cerebrospinal fluid (CSF) (Figure 1A). High-dose corticosteroid treatment seemed to improve her condition. However, ptosis on the left side, headache, and fever became worsened when corticosteroids doses were tapered; therefore, at 35 months of age, she was transferred to the SNUCH. She also had skin rashes on her lower legs. Neurological examination revealed truncal ataxia and limitation of left ocular movement to the upward and medial sides. C-reactive protein (CRP) was elevated up to 9 mg/dL (normal range: 0~0.5 mg/dL) and erythrocyte sedimentation rate (ESR) was elevated up to 75 mm/hr (normal range: 0~20 mm/hr). However, there were no abnormalities in other laboratory tests, such as renal function, liver function, coagulation function, and autoantibodies on blood, and there was no evidence of infection in the blood or CSF. Miller–Fisher syndrome was suspected due to ataxia and ophthalmoplegia, thus intravenous immunoglobulin (IVIG) 2 g/kg was administered. Ataxia improved after IVIG administration, and the patient was discharged.
At 40 months of age, she had colicky abdominal pain with vomiting, fever, and hypertension (150/100 mmHg). Skin necrosis in fingertip (Figure 1B), livedo reticularis and palpable erythematous nodules in both lower extremities (Figure 1C) were observed. Raynaud phenomenon was detected in the fingers and tongue. Raynaud scan showed a remarkable decrease of blood pool (Figure 1D). Computed tomography of the abdomen revealed multiple infarctions in both kidneys (Figure 1E), and a skin biopsy showed superficial perivascular lymphocyte infiltration. Low molecular weight heparin, warfarin, aspirin were commenced as she was suspected of having antiphospholipid antibody syndrome because of a weakly positive lupus anticoagulant test and multiple kidney infarctions. However, the abdominal pain and fever were aggravated. Abdominal angiography revealed multiple aneurysms and peripheral vessels narrowing of the branches of the mesenteric artery, bilateral renal artery, splenic artery, and distal portion of the right coronary and circumferential arteries (Figure 1F). The patient was diagnosed as PAN by satisfying the criteria of livedo reticularis, hypertension, and angiographic abnormality according to the American college of rheumatology (ACR) 1990 criteria for the classification of PAN [9]. There were no specific findings on blood and CSF tests except for elevation of inflammatory markers, such as ESR and CRP, and mild anemia, although various blood tests including autoantibodies and coagulation studies were performed whenever she showed symptoms and signs. In addition, there was no evidence of immunologic and hematologic diseases such as hypogammaglobulinemia and cytopenia. Immunoglobulin G/A/M levels were 1,296/149/207 mg/dL (normal range: 345~1,236, 14~159, 43~207 mg/dL, respectively), WBC count was 7,540 /μL, differential counts were that segmented neutrophil was 34.6%, lymphocyte was 52.3%. Hemoglobin was 11.3 g/dL, Platelet count was 331,000 /μL (normal range: 6,000~15,000 /μL, 10.5~14.0 g/dL, 150,000~450,000 /μL).
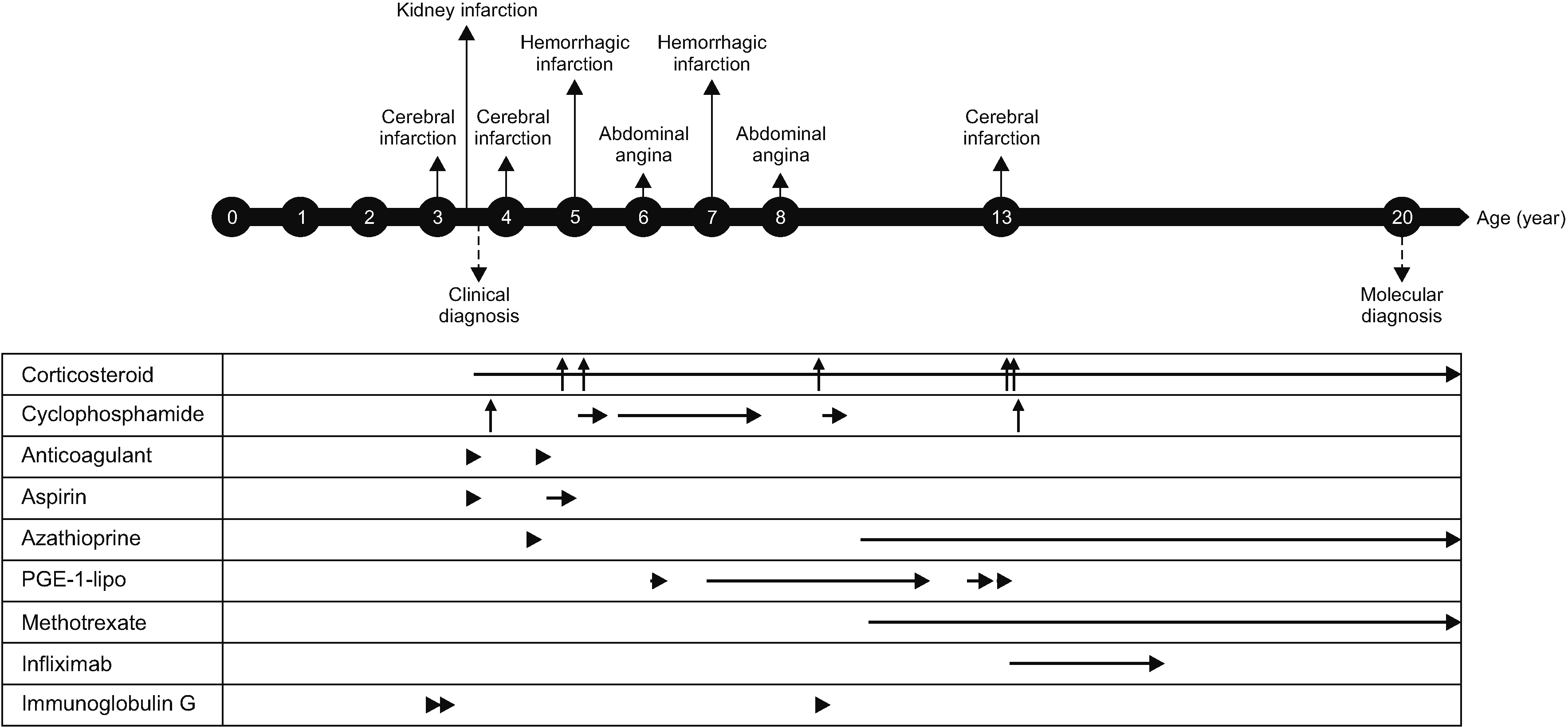
She was treated with corticosteroids, including high-dose methylprednisolone pulse therapy; cytotoxic agents, such as cyclophosphamide; other immunosuppressants such as azathioprine and methotrexate; anticoagulation therapy, such as enoxaparin and aspirin; vasodilators, such as prostaglandin E1; and IVIG. Nonetheless, she had severe intracranial hemorrhage at the age of 5 years that led to right-sided hemiplegia and symptoms and signs caused by vascular inflammation were repeated until 13 years of age (Figure 1G). Finally, a tumor necrosis factor (TNF)-α inhibitor (infliximab) was added at the age of 13 years. TNF-α inhibitor administered 100 mg at intervals of 1 to 2 months and discontinued due to allergic reaction after administration for 2 years. During TNF-α inhibitor administration, azathioprine was reduced from 75 mg to 25 mg, methotrexate from 12.5 mg to 10 mg, and prednisolone from 15 mg (0.5 mg/kg) to 2.5 mg, and her symptoms were not aggravated despite tapering the doses of corticosteroids and immunosuppressants. The patient’s medical events and medications are summarized and expressed in Figure 2.
Recently, we planned molecular analysis as we suspected DADA2 due to her early-onset age, stroke history, and poor response to treatment for PAN. Whole genome sequencing (WGS) was conducted on the patient and her parents, because the first target gene was all regions of the ADA2 gene, including coding and non-coding regions. WGS revealed two different variants of the ADA2 gene (c.753G>A, p.Pro251Pro; c.1069G>A, p.Ala357Thr), which were inherited from both parents. The reference for coding sequence of canonical transcripts is as follows: ADA2 (NM_001282225.2). The first variant, c.753G>A, does not lead to a change in amino acid 251, but it is the last nucleotide of exon 4, which is located in the splice donor site and affects RNA splicing. The other variant, c.1069G>A, has been reported to be a pathogenic variant [10]. Therefore, we performed RNA sequencing to confirm the splicing variant of the ADA2 gene, which showed intron retention on intron 4 and nonsense-mediated mRNA decay on the allele with the splicing variant (Figure 3). Finally, she was diagnosed with DADA2 caused by compound heterozygous pathogenic variants. Now she is doing well with immunosuppressants (prednisolone, azathioprine, methotrexate) without a TNF-α inhibitor.
The study was approved by the Institutional Review Board (IRB) of Seoul National University Hospital (IRB no. H-1606-116-771). The IRB granted a waiver of informed consent.
DISCUSSION
To the best of our knowledge, this is the first report of a case of DADA2 in Korea. She had a biallelic pathogenic variant of the ADA2 gene on chromosome 22, which she inherited from both parents. In addition, we demonstrated abnormal RNA splicing using RNA sequencing, which has been predicted to date.
DADA2 was first described in patients with fever, early-onset stroke, and vasculopathy [4]. Subsequently, various phenotypes have been reported, including immunodeficiency, bone marrow dysfunction, and lymphoid proliferation, such as hepatosplenomegaly. Most DADA2 cases occur in childhood, but adult-onset cases have occasionally been reported [7]. According to a review article that analyzed 316 patients, vasculopathy was the most representative feature, which is similar to PAN [7]. This patient had systemic inflammation and vasculopathy presented as recurrent fever, strokes, multiple kidney infarctions, and livedo reticularis since the age of 34 months, which is consistent with the early-onset vasculopathy phenotype of DADA2 [4,10]. Furthermore, disease activity was not well controlled until the addition of a TNF-α inhibitor, which was supportive of DADA2. The recurrent abdominal pain since the age of 40 months was also thought to be caused by vasculopathy of the gastrointestinal tract, which has been previously reported [5,7]. However, our patient did not have signs of bone marrow dysfunction or immunodeficiency [4–6].
In the treatment of DADA2, immunosuppressive therapies were the mainstay to control systemic inflammation, but its efficacy varies depending on the manifestation or severity of the disease. Although various biological agents have been tested, currently, TNF-α inhibitors are thought to be the treatment of choice. Hematopoietic stem cell transplantation is also recommended in severe cases with hematological or immunological phenotypes [5]. The patient in this study was empirically treated with infliximab (TNF-α inhibitor) for refractory cPAN before confirming the ADA2 variants because several studies have shown that TNF-α inhibitors are effective in stroke prevention and vasculopathy [2,5,7,8,11]. Her symptoms improved with infliximab therapy, and cerebral infarction was no longer experienced with a low dose of immunosuppressive agents despite discontinuing infliximab for over 5 years. Genetic analysis for the ADA2 gene should be conducted in some patients with cPAN to ensure appropriate treatment, including the early administration of TNF-α inhibitors. According to the 2021 ACR/Vasculitis Foundation guideline for the management of PAN, PAN-like syndrome with strokes patients should be considered DADA2, and the use of inhibitors was strongly recommended when the patient was confirmed to be DADA2 [12]. Genetic diagnosis of DADA2 is also recommended for patients with family and early-onset PAN or patients with PAN showing unusual disease course or poor response to conventional therapy [8,13,14]. In the case of our patient, it is consistent with the above conditions in that she also showed early onset cPAN with stroke and poor response to conventional treatment of PAN. Reported studies and this case suggest that ADA2 gene analysis is essential for patients with early-onset cPAN or unusual cPAN with stroke for appropriate therapeutic approaches to DADA2 that can leave permanent complications. The first studies on DADA2 reported that ADA2 enzyme activity in the plasma is significantly decreased in patients with DADA2 [2,4]. In this report, we could not check ADA2 enzyme activity in this patient who had two variants, one of which was a missense variant (c.1069G>A, p.Ala357Thr), and significant loss of ADA2 enzyme activity has already been demonstrated [15]. The other variant (c.753G>A, p.Pro251Pro) was introduced as a splicing variant in a French study, but it was interpreted using in silico tools instead of demonstrating ADA2 enzyme activity [16]. However, we confirmed abnormal protein production in the intron region by splicing variants through RNA sequencing instead of ADA2 enzyme activity (Figure 3). This variant is a silent variant that does not change the amino acid, but it is the last base of the 3’ splice site, exon-intron junction site, on exon 4, which affects aberrant splicing of mRNA, as previously reported [17-19]. In this case, adenine of 753th nucleotide was inherited from her father. However, approximately 33% of adenine was decayed by nonsense-mediated mRNA decay to reduce errors in gene expression, and the remaining 22% of adenine processes mRNA on RNA sequencing called retained intron.
In conclusion, we reported a case of DADA2 in a patient with typical manifestations caused by biallelic variants, including splicing variants. We strongly suggest genetic analysis for the ADA2 gene in patients with early-onset cPAN or cPAN accompanied with stroke to provide prompt and appropriate therapy and to prevent severe complications caused by multiple infarctions.
SUMMARY
This is the first report of a case of DADA2 in Korea, who was diagnosed with cPAN previously. This patient had early-onset stroke and vasculopathy and genetic analysis revealed biallelic variants including splicing variants. We suggest genetic analysis for the ADA2 gene in patients with early-onset cPAN or cPAN accompanied with stroke to provide appropriate therapy and to prevent complications.
 XML Download
XML Download