

 PDF
PDF Citation
Citation Print
Print


INTRODUCTION
Vascular injury initiates infiltration of inflammatory cells into damaged tissues, and causes subsequent release of endogenous damage-associated molecular patterns (DAMPs) including high-mobility group box 1 (HMGB1) [1]. HMGB1 released from activated immune cells or necrotic cells functions as an important inflammatory mediator when present extracellularly [2]. In atherosclerotic plaque, the level of HMGB1 was increased and suggested to be involved in vascular remodeling via the potentiation of inflammatory processes [3,4]. Recent evidences have also indicated that HMGB1 was suggested as a key player in the process of vascular remodeling in the injured vasculatures [5]. Although vascular smooth muscle cells (VSMCs) have been identified as a major source of HMGB1 in atherosclerotic lesions [1,6,7], the precise role played by VSMCs in vascular remodeling in the injured vasculatures is unclear.
When cells are exposed to physical and chemical stresses, the nuclear HMGB1 is translocated outside the cells and then extracellular HMGB1 binds directly to various cell membrane pattern recognition receptors including TLR2, TLR4 and receptors for advanced glycation end-product (RAGE) which were consistently expressed in VSMCs [8-10]. In previous studies, the expression of RAGE in VSMCs at the sites of vascular injury was markedly upregulated compared to that in normal healthy vessels [7,11], thereby enhancing AGE-induced VSMC proliferation [12]. Thus, HMGB1-RAGE interaction has been recognized as a key process involved in the development of vascular remodeling disease [11,13].
The regulation of VSMC proliferation within a normal vascular wall is tightly controlled through the organized balance of several humoral mediators [14,15]. During the atherosclerotic process, the enhanced production of proinflammatory cytokines affects VSMC proliferation, which may contribute to the intimal hyperplasia in atherosclerosis [16,17] Although VSMC proliferation has been implicated as a key process in the progression of vascular remodeling [18,19], the precise role of VSMCs in vascular remodeling in the injured vasculatures is unclear.
RAGE expression is upregulated in the lesion of vascular injury [20] and the increased RAGE expression in VSMCs was indicated as an important player in the development of proliferative vascular diseases [21]. Although the importance of AGE-RAGE interaction in the injured vasculatures has been well established in vascular remodeling [22,23], the intracellular signaling mediating VSMC proliferation remain unclear. Given the importance of mitogen-activated protein kinase (MAPK) signaling in the development of vascular remodeling in the injured vasculatures, the role of MAPK signaling in RAGE expression and AGE-induced cell proliferation in HMGB1-stimulated VSMCs were investigated in this study in order to develop effective therapeutic strategies for vascular remodeling.
METHODS
Chemicals and antibodies
Recombinant human HMGB1 was purchased from R&D Systems, Inc. (Minneapolis, MN, USA). AGE-BSA was purchased from BioVision (Milpitas, CA, USA). PD98059 (an ERK inhibitor), SP600125 (a JNK inhibitor) and SB203580 (a p38 MAPK inhibitor) were purchased from Enzo Life Sciences, Inc. (Ann Arbor, MI, USA). RAGE antibodies were purchased from Abcam (Cambridge, MA, USA). β-Actin antibody was purchased from Santa Cruz Biotechnology, Inc (Beverly, MA, USA). p44/42 MAPK (ERK1/2), SAPK/JNK, p38 MAPK, phospho-p44/42 MAPK (ERK1/2) (Thr202/Tyr204), phospho-SAPK/JNK (Thr183/Tyr185) and phospho-p38 MAPK (Thr180/Tyr182) antibodies were purchased from Cell Signaling Technology (Beverly, MA, USA). Horseradish peroxidase (HRP)-conjugated IgG antibody (Santa Cruz Biotechnology, Inc.) was used as the secondary antibody.
Cell culture
Human aortic VSMCs (hVSMCs) purchased from the ATCC (Manassas, VA, USA) were grown in culture dishes using smooth muscle cell growth medium (Gibco BRL, Grand Island, NY, USA), smooth muscle growth supplement (Gibco BRL), 10% fetal bovine serum, antibiotic-antimycotic solution (Gibco BRL). Cells were incubated at 37°C in a humidified atmosphere containing 5% CO2/95% air. hVSMCs between passages 4 and 8 were used for this experiment.
Cell proliferation assay
hVSMC proliferation rates were determined by using an MTT assay. In brief, hVSMCs (1 × 105 cells) were treated with a MTT working solution (EZ-Cytox; Daeil Laboratories, Seoul, Korea), and then incubated in a humidified atmosphere containing 5% CO2/95% air for 1 h at 37°C. For ELIZA analysis, the absorbance value of the solution was obtained at a wavelength of 450 nm. Relative proliferation rates were determined by comparing cells with control.
RNA isolation and RT-PCR
The expression of RAGE mRNA in hVSMCs was determined by RT-PCR using GAPDH as an internal control. In accordance with the manufacturer’s instructions, total RNA was isolated using Trizol reagent (Invitrogen, Carlsbad, NY, USA). RNA (1 μg) was reverse transcribed into cDNA using the ImProm-II reverse transcription system (Promega, Madison, WI, USA). PCR amplification was carried out using RAGE-specific primers (forward, 5´-GC TGT CAG CAT CAG CAT CAT-3´ reverse, 5´-AT TCA GTT CTG CAC GCT CCT-3´). Equal amounts of RT-PCR products obtained were separated on 1%–1.5% agarose gel and then stained with ethidium bromide. Signals from bands were quantified using the UN-SCAN-IT GEL 7.1 program and data were expressed as relative GAPDH densities.
Western blot analysis
hVSMC lysates were prepared in an ice-cold lysis buffer (Thermo Fisher Scientific, Waltham, MA, USA). Equal amounts of protein were separated on 8%–10% sodium dodecyl sulfate-polyacrylamide gels and then transferred to nitrocellulose membranes (Amersham-Pharmacia Biotech, Piscataway, NJ, USA). Membranes were blocked with 5% skim milk in Tris buffered saline with Tween-20 (TBST) at room temperature for 2 h and then incubated overnight with primary antibody at 4°C. Following this, membranes were washed with TBST, incubated with horseradish peroxidase-conjugated secondary antibody for 2 h at room temperature and then chemiluminescence intensities were measured using image capturing software (Amersham, Imager 680 version. 2.0). Membranes were re-blotted using anti-β-Actin antibody as an internal control. Signals from bands were quantified using the UN-SCAN-IT GEL 7.1 program and data were expressed as relative β-Actin densities.
Small interfering RNA (siRNA) preparation and transfection
siRNA for p44/42 MAPK (ERK1/2) (Cat # 6560), SAPK/JNK (Cat # 6232) and p38 MAPK (Cat # 6564) were purchased from Cell Signaling Technology. The siRNA negative control duplex was used as a control oligonucleotide. For siRNA transfection, hVSMCs were seeded and then transfected with MAPKs siRNA using Lipofectamine 2000 reagent (Invitrogen) according to the manufacturer’s protocol.
Statistical analysis
Results were expressed as the means ± standard error of the means (SEMs). One-way analysis of variance (ANOVA) followed by Dunnett multiple comparison test was performed. Student’s t-test was used to determine significant differences between two groups. Statistical significance was accepted for p-values < 0.05.
RESULTS
Characteristics of RAGE expression in hVSMCs stimulated with HMGB1
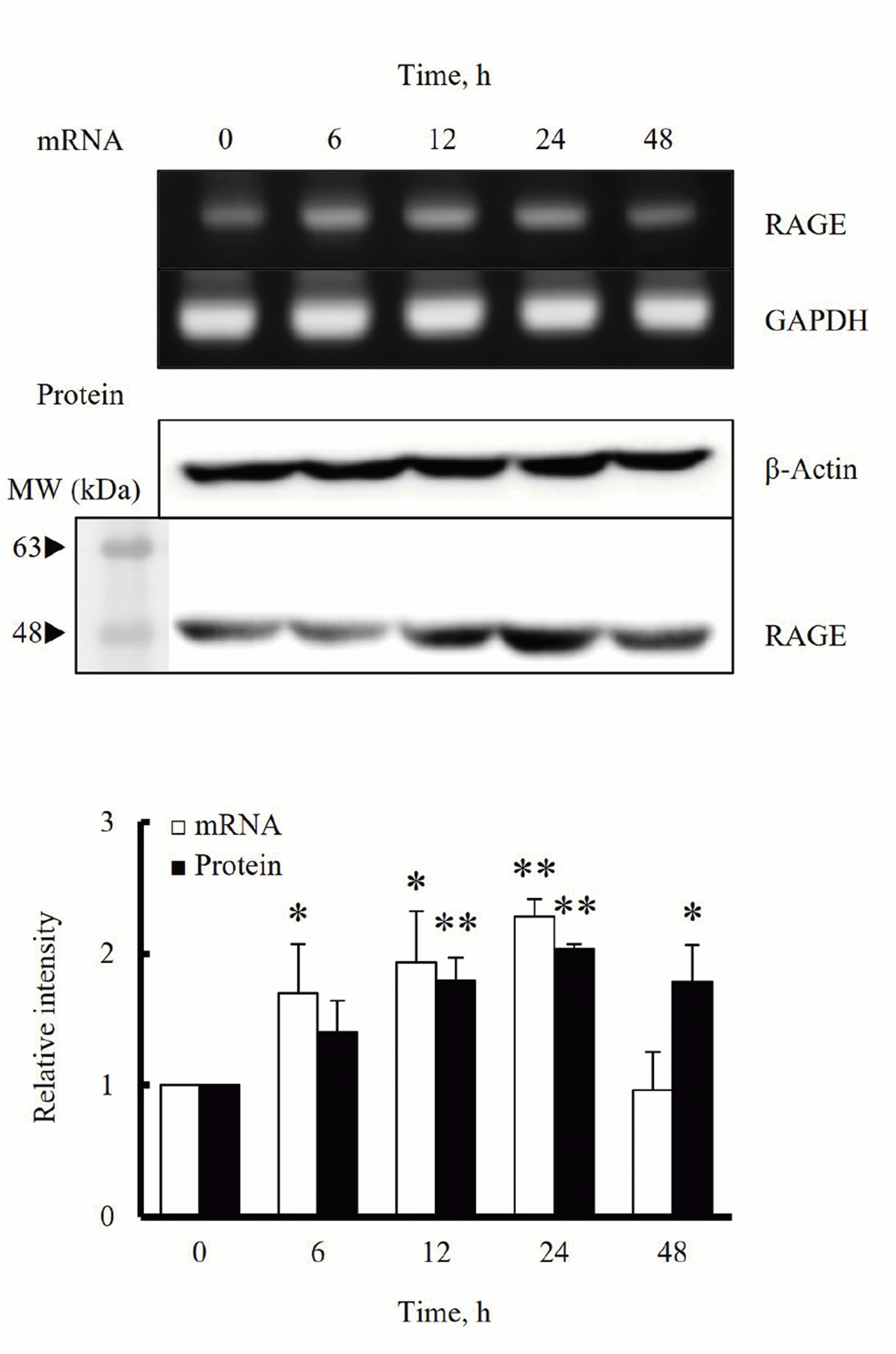
To investigate the effects of HMGB1 on RAGE expression in hVSMCs, the expression of RAGE mRNA and protein in cells stimulated with HMGB1 was determined by using RT-PCR and Western blot, respectively. In cultured hVSMCs stimulated with HMGB1 (100 ng/ml) for 0 to 48 h, the expression of RAGE mRNA started to increase at 6 h of HMGB1 stimulation, and then continuously increased until 24 h. Likewise, the RAGE protein expression in cells stimulated with HMGB1 started to increase at 12 h stimulation, and then peaked at 24 h after HMGB1 treatment (Fig. 1).
Functional role of HMGB1-induced RAGE in hVSMC proliferation by AGE
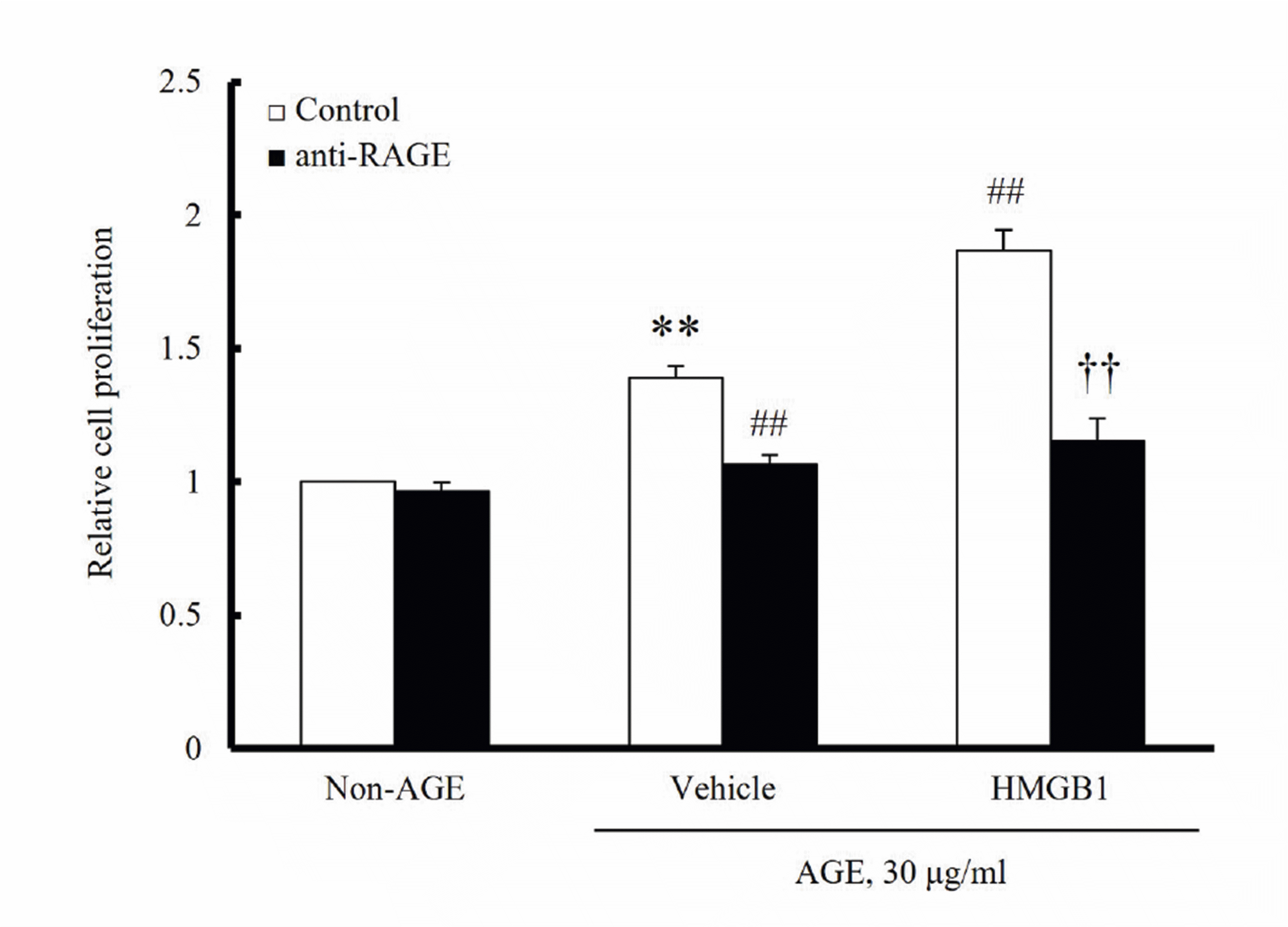
To increase RAGE expression in VSMCs, hVSMCs were treated with HMGB1 (100 ng/ml) for 24 h. Cell proliferation was induced by AGE and then measured using an MTT assay. As shown in Fig. 2, AGE (30 μg/ml)-induced cell proliferation was markedly increased in VSMCs stimulated with HMGB1 (100 ng/ml) compared to vehicle-treated cells. The inhibitory effects of anti-RAGE antibody on the AGE-induced proliferation in HMGB1-treated cells was more prominent than that in vehicle group. Thus, the RAGE signaling upregulated by HMGB1 was suggested as a key player in VSMC proliferation in the injured vasculatures.
Characteristics of MAPK phosphorylation in HMGB1-stimulated hVSMCs
To determine the potential participation of MAPK signaling in VSMC proliferation in the injured vasculatures, this study determined the time-dependent patterns of MAPK phosphorylation in VSMCs stimulated with HMGB1. In this study, hVSMCs were treated with HMGB1 (100 ng/ml) for 0 to 24 h, and then the levels of phosphorylated and total MAPK subfamilies were determined by Western blotting. As shown in Fig. 3A, the increase in ERK phosphorylation in cells stimulated with HMGB1 was peaked at 6 h, and maintained high levels until 24 h. The phosphorylation of JNK and P38 MAPK was time-dependently increased until 24 h after HMGB1 stimulation (Fig. 3B, C).
The individual role of MAPKs subfamilies on RAGE expression in HMGB1-stimulated hVSMCs
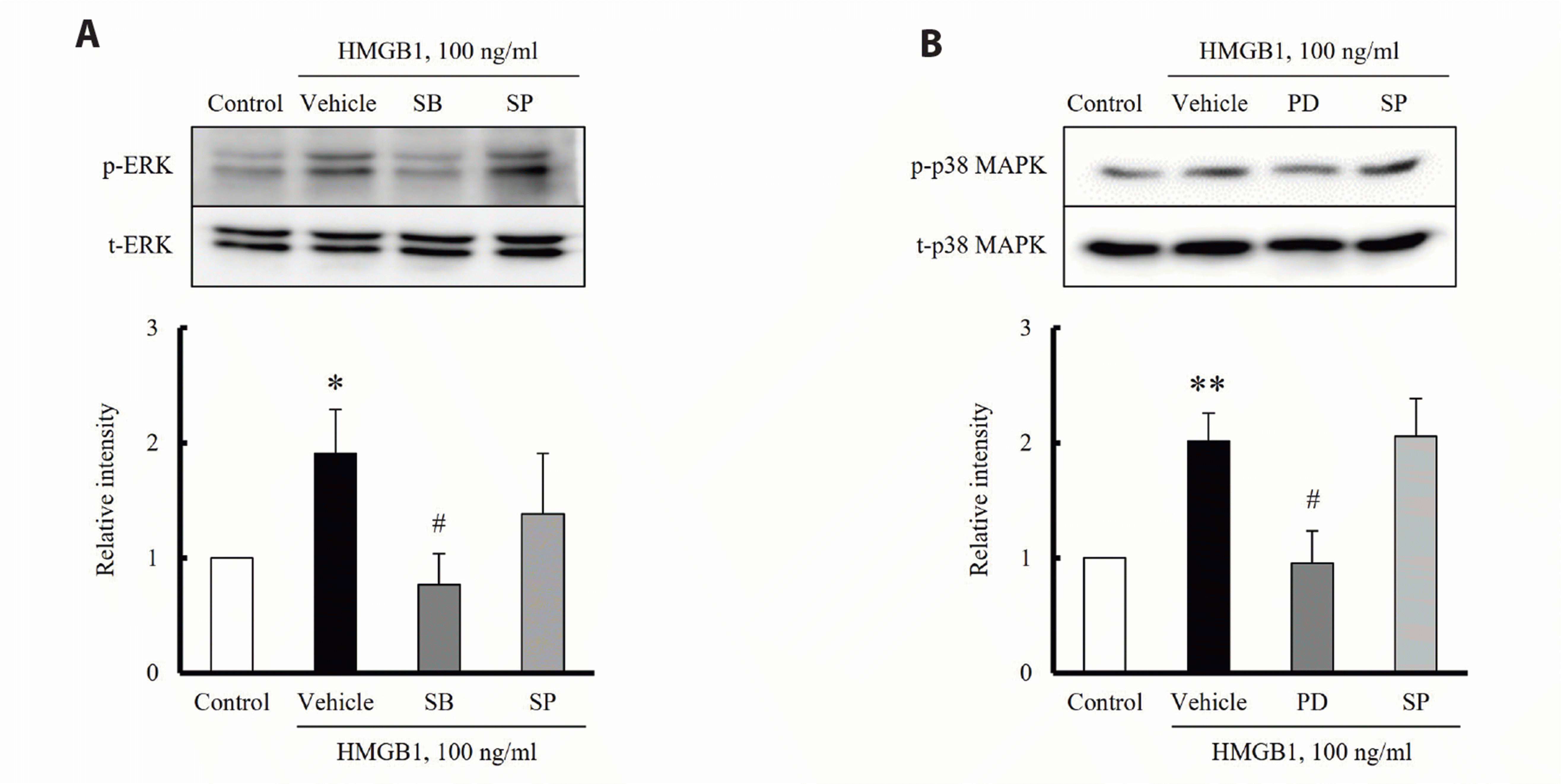
To determine the individual roles of MAPK subfamilies on HMGB1-induced RAGE expression in hVSMCs, RAGE expression was induced by HMGB1 in the presence of various MAPK inhibitors including inhibitors for ERK (PD98059, 10 μM), JNK (SP600125, 10 μM) and p38 MAPK (SB203580, 10 μM). As shown in Fig. 4, the HMGB1-induced RAGE expression was attenuated in cells pretreated with inhibitors for ERK and p38 MAPK, but not in cells treated with a JNK inhibitor. Likewise, HMGB-1-induced RAGE expression was significantly attenuated in cells deficient of ERK and p38 MAPK using their specific siRNAs, but not in JNK-deficient cells, suggesting a pivotal role of ERK and p38 MAPK signaling in RAGE expression in HMGB1-stimulated cells (Fig. 5).
To further determine crosstalk among ERK, JNK, and p38 MAPK signaling, the HMGB1-induced phosphorylation of ERK and p38 MAPK was determined in cells pretreated with various inhibitors for MAPKs. When VSMCs were stimulated with HMGB1 in the presence of p38 MAPK and JNK inhibitors, ERK phosphorylation was inhibited by SB203580 (10 μM), a P38 MAPK inhibitor, but not by JNK inhibitor. Also, the phosphorylation of p38 MAPK was attenuated by PD98059 (10 μM), an ERK inhibitor, but not by JNK inhibitor, suggesting a link between ERK and p38 MAPK signaling on RAGE expression in the HMGB1-stimulated cells (Fig. 6).
The role of ERK/p38 MAPK signaling in HMGB1-augmented effects on AGE-induced VSMC proliferation
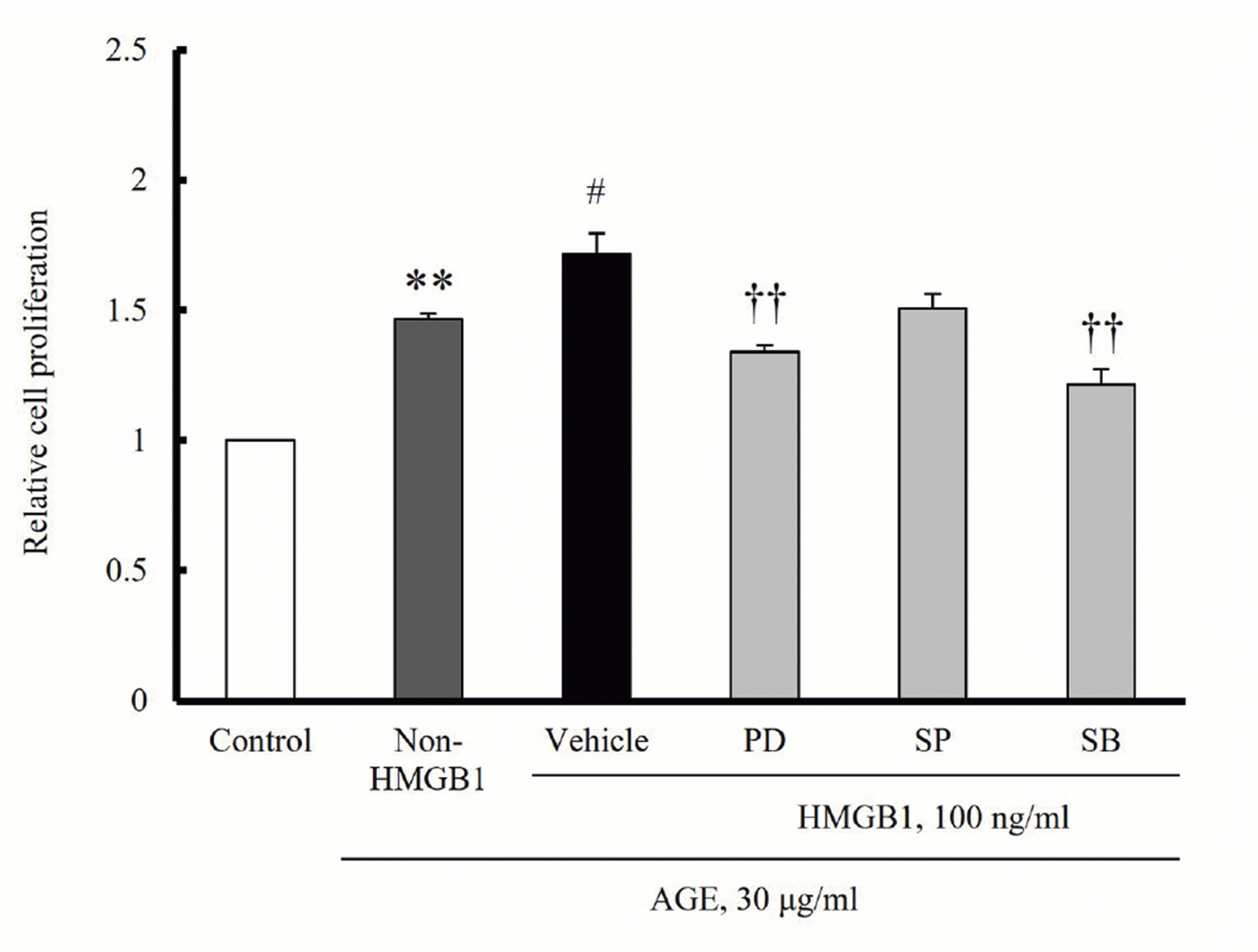
To further confirm whether ERK and p38 MAPK signalings are involved in the augmented proliferative effects of HMGB1 on AGE-stimulated hVSMCs, cells were treated with HMGB1 (100 ng/ml) for 24 h, and then cell proliferation was induced by AGE. In hVSMCs stimulated with HMGB1, AGE-induced cell proliferation was markedly increased, which was attenuated in cells pretreated with PD98059 (10 μM), an ERK inhibitor and SB203580 (10 μM), a p38 MAPK inhibitor, but not by a JNK inhibitor (Fig. 7). These results suggested a pivotal role of ERK and p38 MAPK signaling in regulating RAGE expression and subsequent VSMC proliferation induced by AGE.
DISCUSSION
The salient findings of this study are that the expression of RAGE mRNA and protein was increased in human VSMCs stimulated with HMGB1, which was accompanied by an augmented cell proliferation induced by AGE. Both HMGB1-induced RAGE expression and HMGB1-augmented effects of AGE-induced VSMC proliferation were markedly attenuated by inhibition of ERK and p38 MAPK signaling, suggesting a potential involvement of the ERK/p38 MAPK-RAGE signaling axis in vascular remodeling in the injured vasculatures.
The regulation of VSMC proliferation within a normal vascular wall is tightly controlled through the organized balance of various humoral mediators [14,15]. During the atherosclerotic process, the enhanced production of proinflammatory cytokines affects VSMC proliferation, which may contribute to intimal hyperplasia in atherosclerosis [16,17]. Among various cells in blood vessel walls, VSMCs are the major cell type and an increased VSMC proliferation is implicated in the pathogenesis of vascular remodeling [18,19]. Although VSMC has been suggested as a key player in vascular remodeling in the injured vasculatures, the precise molecular mechanisms involved in VSMC proliferation are unclear.
Vascular injury-induced infiltration of inflammatory cells into damaged tissues is followed by an increased production of endogenous DAMPs including HMGB1, a best characterized and pivotal DAMPs released into the injured vasculatures [1]. In atherosclerotic lesions, VSMCs, macrophage, and endothelial cells were identified as major sources of HMGB1 production [1,6,7]. The increased HMGB1 in atherosclerotic plaque is known to be involved in vascular remodeling via the potentiation of inflammatory processes [3,4]. Recent evidences have also indicated that HMGB1 released from the injured vasculatures was suggested as a key player mediating vascular remodeling [5]. Although VSMCs have been identified as a major vascular source of HMGB1 in atherosclerotic lesions [1,6,7], the precise role of HMGB1 on VSMC proliferation remains unidentified.
HMGB1 primarily resides in the nucleus of quiescent cells [24], and its release is increased when cells are activated in the injured tissues [1,6]. When cells are exposed to physical and chemical stresses, nuclear HMGB1 can be translocated to cytosol and then released outside the cells via active and passive pathways, and the extracellular HMGB1 may bind to cell surface receptors [8,9]. Among various cell membrane pattern recognition receptors, TLR2, TLR4, and RAGE were consistently expressed in VSMCs in our previous study [10]. In diabetes, the increasing evidence shows that RAGE is recognized as an important molecule involved in the development of vascular remodeling disease [13]. Also, the expression of RAGE in VSMCs at the sites of vascular injury was markedly upregulated compared to that in normal healthy vessels [7,11], thereby enhancing AGE-induced VSMC proliferation [12]. To investigate the mechanisms involved in HMGB1-mediated vascular remodeling, we investigated the regulatory effects of HMGB1 on the expression of RAGE in human VSMCs. In the present study, stimulation of VSMCs with HMGB1 increased RAGE expression, which was accompanied by an augment in AGE-induced cell proliferation. Thus, the HMGB1-RAGE signaling in hVSMCs was suggested as a pivotal pathway involved in vascular remodeling in the injured vasculature.
The MAPK signaling is activated in the injured vasculatures, and the activation of MAPK pathways is shown to be involved in the exaggerated neointimal hyperplasia after balloon injury [25]. In angiotensin II-induced VSMC proliferation, the importance of the increased phosphorylation of ERK1/2, JNK1/2, and p38 MAPK has been demonstrated [26]. Given the importance of MAPK signaling in the neointima hyperplasia in the injured vasculatures, we investigated the role of HMGB1 on the regulation of MAPK activities. In line with other previous study in which HMGB1 activates ERK1/2 and p38 MAPK in endothelial cells [27], the phosphorylation of ERK, JNK and p38 MAPK was increased in hVSMCs treated with HMGB1 in our present study. The increase in ERK phosphorylation in cells stimulated with HMGB1 was peaked at 6 h, and maintained high levels until 24 h. The phosphorylation of JNK and p38 MAPK was time-dependently increased until 24 h after HMGB1 stimulation. Typically, the activation of MAPK is an early event in cellular activation, thus continuous increment of MAPK phosphorylation might be caused by secondary activation by other stimuli including humoral factors. The long-term phosphorylation of MAPK signaling in our present study should be confirmed by future experiments.
The increased RAGE expression has been suggested as a key player in the progression of vascular remodeling. However, the precise mechanisms regulating RAGE expression in VSMCs in the injured vasculatures is unclear. To investigate the importance of MAPK signaling in an increased VSMC proliferation in the injured vasculatures, this study determined the role of MAPK signaling in HMGB1-induced RAGE expression. In cultured hVSMCs stimulated with HMGB1, RAGE mRNA and protein expression were markedly increased, which was attenuated in cells pretreated with inhibitors of ERK and p38 MAPK as well as in cells deficient of ERK and p38 MAPK using siRNAs, but not in cells deficient of JNK. In line with an increased RAGE expression in HMGB1-stimulated cells, VSMC proliferation induced by AGE was markedly augmented in cells stimulated with HMGB1, which was attenuated in cells pretreated with ERK and p38 MAPK inhibitors. Thus, it was suggested that ERK and p38 MAPK activated by HMGB1 mediate RAGE expression in VSMCs, which augments AGE-mediated VSMC proliferation.
To further determine crosstalk among ERK, JNK, and p38 MAPK signaling in vascular remodeling, the HMGB1-induced phosphorylation of ERK and p38 MAPK was determined in cells pretreated with various inhibitors for MAPKs. When VSMCs were stimulated with HMGB1 in the presence of p38 MAPK and JNK inhibitors, ERK phosphorylation was inhibited by a p38 MAPK inhibitor, but not by JNK inhibitor. Also, the phosphorylation of p38 MAPK was attenuated by an ERK inhibitor, but not by JNK inhibitor, suggesting a link between ERK and 38 MAPK signaling in HMGB1-stimulated cells.
Taken together, the results of our present study show that the activation of ERK and p38 MAPK signaling mediates RAGE expression in HMGB1-stimulated hVSMCs, which augments proliferation of human VSMC induced by AGE. Considering the importance of the phenotypic modulation of VSMCs in vascular remodeling, the HMGB1-ERK/p38 MAPK-RAGE signaling axis in VSMCs was suggested as a potential therapeutic target for vascular remodeling in the injured vasculatures.

 XML Download
XML Download