

 PDF
PDF Citation
Citation Print
Print


INTRODUCTION
Drug repositioning of approved therapies might extend their therapeutic potential against resistant cancer cells [1]. Benzimidazole anthelmintic agents are relatively non-toxic to normal cells [2-6] with a half-maximal inhibitory concentration (IC50) of 5 µM for less sensitivity in 461 cancer cells [7]. A recent report summarized the anti-tumourigenic activity of benzimidazole as follows [1]: 1) cell cycle arrest at G2/M phase with increased levels of cyclin B1, p21 and p27Kip1; 2) apoptosis with increased expression of caspase-3, poly (ADP-ribose) polymerase (PARP), and cytochrome-C; 3) autophagy with increased microtubule-associated protein 1A/1B-light chain 3 (LC3) and Beclin-1; and 4) altered cell viability or differentiation with increased p53, p21, p38, and c-Jun N-terminal kinase (JNK), and decreased extracellular signal-regulated protein kinase (ERK).
Self-administration of fenbendazole, a benzimidazole anthelmintic agent, by patients with cancer has been reported in social media [8,9]. Although anti-cancer effects of fenbendazole as an alternative or supplementary agent were recently reported in a case series of genitourinary malignancies [10], no definitive evidence of anti-cancer effects exists in human because of its toxicity and teratogenicity [8]. Nevertheless, the potential for drug repositioning of fenbendazole requires further investigation because of the much higher IC50 values in normal cells compared with cancer cells [5].
Colorectal cancer (CRC) is the third most common cancer diagnosed and the second leading cause of cancer death worldwide [11]. Although 5-fluorouracil (5-FU) remains the mainstay of standard chemotherapy for CRC, various resistance mechanisms are reported [12]. Especially, mutations in p53 [13-15] or p38α mitogen-activated protein kinase (MAPK) [16,17] are associated with chemo-resistant CRC. To overcome the resistance, activation of transforming growth factor-β [18] or ERK [19] was suggested in 5-FU-resistant CRC cell line. Upregulation of protein kinase B (Akt) is a critical factor in the progression of CRC [13]. It induces apoptosis via inhibition of rapidly accelerated fibrosarcoma (a family of three serine/threonine-specific protein kinases) – ERK activation [20].
In terms of benzimidazole, Nygre et al. [21] screened a compound library to identify agents with potential for repositioning in cancer therapy, and found that mebendazole and albendazole have anti-cancer activity in 80% of CRC cell lines. Benzimidazole inhibited the proliferation of HCT116 cells [22] and induced apoptosis via activated JNK in HCT116 and SW480 cells [23]. Flubendazole induced mitotic catastrophe with increased cyclin B1 and caspase-3-PARP pathway [24]. It has an anti-migratory effect by inhibiting the expression of nuclear factor kappa-light-chain-enhancer of activated B cells p65 in CRC cells derived from primary tumor and lymph node metastasis [25]. In addition, Williamson et al. [26] showed that mebendazole combined with non-steroidal anti-inflammatory drugs reduced tumor initiation by decreasing c-Myc and increasing apoptosis in an ApcMin/+ mouse model of familial adenomatous polyposis.
Although fenbendazole enhance the cytotoxicity of radiation or docetaxel and increased the anti-cancer effects of radiation against mammary tumors [27], only a few studies evaluated at the anti-cancer effects of benzimidazole against metastatic or resistant cancers [28-30]. Albendazole effectively inhibited paclitaxel- and doxorubicin-resistant lung cancer cells [31] and thereby reduced multidrug resistance. Nonetheless, the role of benzimidazole in resistant CRC has yet to be reported. Therefore, we aimed to evaluate the effects and underlying mechanisms of fenbendazole in SNU-C5/5-FUR cells as compared with wild type SNU-C5 cells in order to identify therapeutic strategies to overcome resistance to 5-FU.
METHODS
Reagents and antibodies
MTT was purchased from Amresco, Inc. (VWR International LLC, Seongnam, Korea). 5-fluorouracil (5-FU, #F6627), albendazole (#PHR 1281), deferoxamine mesylate (DFOM, #D9533), fenbendazole (#PHR 1832), and ferrostatin-1 (#SML 0583) were obtained from Sigma-Aldrich (Merck KGaA, Darmstadt, Germany).
Antibodies specific for c-Myc (9E10; #sc-40; diluted 1:1,000), cytochrome-C (H-104; #sc-7159; diluted 1:1,000), ERK (C-16; #sc-93; diluted 1:2,000), ferritin heavy chain (B-12) (FTH1; #sc-376594; diluted 1:2,000), GAPDH (#sc-47724; diluted 1:2,000), and phospho-p53 (Ser6; #sc135630; diluted 1:1,000), transferrin receptor (H68.4) (TfRC; #sc-65882; diluted 1:2,000) were purchased from Santa Cruz Biotechnology (Santa Cruz, CA, USA); glutathione peroxidase 4 (GPX4 [EPNCIR144]; #ab125066; diluted 1:1,000) and SLC40A1 (ferroportin, FPN; #ab78066; diluted 1:1,000) were purchased from Abcam (Cambridge, UK); caspase-3 (#9665; diluted 1:1,000), cleaved caspase-3 (#9661; diluted 1:1,000), cyclin B1 (#4138; diluted 1:1,000), phospho-ERK (Thr202/Tyr204; #4370; diluted 1:2,000), JNK (#9252; diluted 1:1,000), phosphor-JNK (Thr183/Tyr185) (#9251; diluted 1:1,000), p21 (#2947; diluted 1:1,000), p27 (#2552; diluted 1:1,000), p38 MAPK (#9212; diluted 1:1,000), phospho-p38 MAPK (Thr180/Tyr182; #9211; diluted 1:1,000), PARP (#9542; diluted 1:1,000), autophagy antibody sampler kit (#4445; consisting of Beclin-1 (D40C5), LC3 (D3U4C), Atg5 (D5F5U), Atg12 (D88H11), Atg16L1 (D6D5), Atg7 (D12B11), and Atg3; diluted 1:2,000/each), and necroptosis antibody sampler kit (#98110; consisting of receptor-interacting protein kinase (RIP) (D94C12), phospho-RIP (Ser166) (D1L3S), mixed lineage kinase domain-like protein (MLKL) (D2I6N), phospho-MLKL (Ser358) (D6H3V), receptor-interacting protein kinase 3 (RIP3) (E1Z1d), and phospho-RIP3 (S227) (D6W2T); diluted 1:1,000/each) were purchased from Cell Signaling Technology Inc. (Beverly, MA, USA); the high mobility group box 1 (HMGB1; #GTX62170; diluted 1:2,000) and p53 (#GTX70218; diluted 1:1,000) were purchased from GeneTex (Irvine, CA, USA); SLC7A11 (cysteine/glutamate transporter ; #ANT-111; diluted 1:2,000) was purchased from Alomone Labs (Jerusalem, Israel).
Cell culture and viability assay
SNU-C5 (IC50 against 5-FU = 5 μM) and the 5-fluorouracil-resistant SNU-C5 (SNU-C5/5-FUR; IC50 against 5-FU = 140 μM) cell lines were cultured as previously described [19,32].
To achieve similar confluency after 3 days of incubation, SNU-C5 (2 × 103 cells/well) and SNU-C5/5-FUR (5 × 103 cells/well) cells were seeded in 96-well plates. Cells were treated with benzimidazole (fenbendazole and albendazole) and anti-ferroptotic agents (ferrostatin-1 and DFOM) at various concentrations. The effect of drugs on cell viability was evaluated via reduction of MTT to its formazan product with a VERSAmax microplate reader (Molecular Devices Korea LLC, Seoul, Korea) as previously reported [19].
Flow cytometry
Cells were treated with or without fenbendazole (0.5 and 5.0 μM for SNU-C5 and SNU-C5/5-FUR cells, respectively) for 3 days, followed by flow cytometry using the FACSCalibur system (BD Biosciences, San Jose, CA, USA) and BD FACStation software version 6.0 (BD Biosciences) as described previously [19,32].
Statistical analysis
All data were compiled from a minimum of three replicate experiments. The data are expressed as mean values ± SD. Statistically significant differences (p < 0.05) were found using Student’s paired t-test or one-way analysis of variance (ANOVA) with a Bonferroni post-hoc test (SPSS version 14.0; SPSS Inc., Chicago, IL, USA).
RESULTS
Benzimidazole induced dose- and time-dependent anti-proliferative effects
Fenbendazole and albendazole showed dose-dependent anti-proliferative effects against both SNU-C5 and SNU-C5/5-FUR cells (Fig. 1A, C). After 3 days of incubation, the IC50 values of the SNU-C5 cells treated with fenbendazole and albendazole were 0.50 and 0.47 μM, respectively. The IC50 values of SNU-C5/5-FUR cells treated with fenbendazole and albendazole were 4.09 and 4.23 μM, respectively. Cell viability of SNU-C5 cells was s 42.24 ± 2.71% and 38.18 ± 2.01% when incubated with 1 μM of fenbendazole and albendazole, respectively. Cell viability of SNU-C5/5-FUR cells was 65.66 ± 1.83% and 67.57 ± 1.58% when treated with 1 μM of fenbendazole and albendazole, respectively, which was further decreased to 30.79 ± 2.35% and 39.78 ± 1.40% at 10 μM concentrations.
Fenbendazole was more effective than albendazole against SNU-C5/5-FUR cells, and time-dependent anti-cancer effects of fenbendazole were further evaluated (Fig. 1B, C). Fenbendazole showed time-dependent anti-proliferative effects on both CRC cells. During 3 days of incubation, the proliferation of SNU-C5 cells increased 4.07 ± 0.18-fold with vehicle and 1.63 ± 0.11-fold with 1 μM of fenbendazole when compared with vehicle-treated control. During 3 days of incubation, SNU-C5/5-FUR cells proliferated 4.60 ± 0.41-fold with vehicle, and 2.94 ± 0.25 and 1.44 ± 0.15-fold with 1 μM and 10 μM of fenbendazole, respectively, when compared with vehicle-treated control. Taken together, a dose of close to the IC50 values of fenbendazole, 0.5 μM for SNU-C5 cells and 5 μM for SNU-C5/5-FUR cells, was used in subsequent experiments.
Fenbendazole induced cell cycle arrest at G2/M phase with increased p21 expression on CRC cells
Cell cycle analysis resulted in a significant increase in the fraction of G2/M phase (Fig. 2A). The fraction of SNU-C5 (12.50 ± 0.92% vs. 21.07 ± 1.10%) and SNU-C5/5-FUR (24.24 ± 0.28% vs. 84.30 ± 1.66%) cells in the G2/M phase showed a significant upward trend, while the fraction of SNU-C5 (56.40 ± 0.69% vs. 52.20 ± 0.38%) and SNU-C5/5-FUR (39.53 ± 0.39% vs. 3.55 ± 0.13%) cells in the G0/G1 phase showed a significant downward trend. The proportion of S phase considerably decreased in SNU-C5 (29.91 ± 0.30% vs. 25.12 ± 0.77%) and SNU-C5/5-FUR (36.23 ± 0.24% vs. 12.15 ± 1.61%) cells.
Western blot revealed that p27 showed a significant increase in the number of SNU-C5 (2.82 ± 0.04-fold) and SNU-C5/5-FUR (2.43 ± 0.17-fold) cells. P21 expression was significantly increased in SNU-C5 (1.98 ± 0.07-fold) and SNU-C5/5-FUR (1.68 ± 0.13-fold) cells. Levels of c-Myc decreased considerably in SNU-C5 (0.59 ± 0.07-fold) and SNU-C5/5-FUR (0.72 ± 0.01-fold) cells, while cyclin B1 levels did not change considerably (Fig. 2B).
Fenbendazole induced apoptosis via caspase-3-PARP pathway in CRC cells
The cell death effects were identified via annexin V/PI staining after incubation with fenbendazole in SNU-C5 and SNU-C5/5-FUR cells (Fig. 3A). Cell death analysis showed a significant increase in the fraction of apoptosis and necrosis was found in both CRC cells compared with vehicle-treated cells (Fig. 3A). The fraction of SNU-C5 cells in early (1.24 ± 0.15% vs. 14.34 ± 0.26%) and late (3.95 ± 0.14% vs. 14.30 ± 0.08%) apoptosis along with the fraction of SNU-C5/5-FUR cells in early (0.25 ± 0.03% vs. 3.27 ± 0.14%) and late (1.83 ± 0.07% vs. 10.77 ± 0.72%) apoptosis showed a significant upward trend. The fraction of SNU-C5 (2.05 ± 0.09% vs. 7.59 ± 0.36%) and SNU-C5/5-FUR (1.40 ± 0.08% vs. 3.16 ± 0.16%) cells in necrosis also showed a significant upward trend.
Western blot revealed that the activation (cleaved/full length) of PARP was significantly increased in SNU-C5 (1.79 ± 0.11-fold) and SNU-C5/5-FUR (2.28 ± 0.03-fold) cells. The activation (cleaved/total form) of caspase-3 was significantly increased in SNU-C5 (1.44 ± 0.10-fold) and SNU-C5/5-FUR (2.18 ± 0.09-fold) cells. The level of cytochrome-C showed a significant increase in SNU-C5 (1.37 ± 0.17-fold) and SNU-C5/5-FUR (1.37 ± 0.08-fold) cells (Fig. 3B).
Fenbendazole did not activate other cell death pathways in CRC cells
Western blot on autophagy proteins revealed that Beclin-1 expression was significantly increased in SNU-C5 (1.35 ± 0.02-fold) and SNU-C5/5-FUR (1.46 ± 0.07-fold) cells. LC3-I level increased slightly in SNU-C5 (1.13 ± 0.13-fold) cells but remained unchanged in SNU-C5/5-FUR cells. Atg7 expression increased considerably in SNU-C5 (1.53 ± 0.07-fold) and in SNU-C5/5-FUR (1.12 ± 0.06-fold) cells. Atg16L1 levels increased slightly in SNU-C5/5-FUR (1.21 ± 0.04-fold) but remained unchanged in SNU-C5 cells. Other markers were not changed considerably in both CRC cells (Fig. 4).
Western blot on ferroptosis proteins revealed that GPX4 showed significantly decreased levels in SNU-C5 (0.60 ± 0.02-fold) and SNU-C5/5-FUR (0.71 ± 0.05-fold) cells. FTH1 levels increased significantly in SNU-C5 (5.44 ± 0.32-fold) and SNU-C5/5-FUR (3.48 ± 0.54-fold) cells. FPN showed a significant increase in SNU-C5 (1.77 ± 0.07-fold) and SNU-C5/5-FUR (1.59 ± 0.10-fold) cells. The levels of SLC7A11 increased slightly in SNU-C5 (1.19 ± 0.05-fold) cells but decreased significantly in SNU-C5/5-FUR (0.67 ± 0.02-fold) cells. The expression of transferrin receptor remained unchanged in both CRC cells. HMGB1, a member of damage-associated molecular patterns (DAMP), showed significantly increased levels in SNU-C5 (1.30 ± 0.10-fold) and SNU-C5/5-FUR (2.36 ± 0.06-fold) cells (Fig. 5A).
The effects of anti-ferroptotic agents, ferrostatin-1 and DFOM, were evaluated in fenbendazole-induced ferroptosis (Fig. 5B). Ferrostatin-1 co-treatment did not increase the viability of both CRC cells. Concentrations greater than 1 μM paradoxically decreased the cell viability in SNU-C5 (69.60 ± 4.85% vs. 47.10 ± 1.71%) and SNU-C5/5-FUR (75.95 ± 2.20% vs. 49.97 ± 1.15%) cells following co-treatment. Co-treatment with DFOM did not increase the viability of the CRC cells. High concentration (10 μM) of DFOM paradoxically decreased the viability of SNU-C5 (60.14 ± 2.11% vs. 35.33 ± 1.10%) and SNU-C5/5-FUR (73.04 ± 1.96% vs. 60.58 ± 1.61%) cells.
Western blot on necroptotic proteins revealed that the activation (phosphor/total) of RIP was significantly increased in SNU-C5 (2.31 ± 0.21-fold) and SNU-C5/5-FUR (4.72 ± 0.32-fold) cells. The activation of RIP3 was significantly decreased in SNU-C5 (0.42 ± 0.00-fold) and SNU-C5/5-FUR (0.47 ± 0.04-fold) cells. The activation of MLKL was not changed considerably in both CRC cells (Fig. 6). While the full length of caspase-8 was not changed in both CRC cells, the significant increase was observed in the active cleaved fragment in SNU-C5 (2.45 ± 0.21-fold) cells and the cleaved intermediate fragment in SNU-C5/5-FUR (1.22 ± 0.07-fold) cells (Fig. 6).
Fenbendazole activated p53 on SNU-C5 cells, but not on SNU-C5/5-FUR cells
The activation (phosphor/total form) of p53 was significantly increased in SNU-C5 (1.17 ± 0.05-fold) cells, but the activation of p38, ERK, and JNK did not change. The activation of p53, p38 (0.83 ± 0.02-fold), ERK, and JNK showed no significant changes in SNU-C5/5-FUR cells (Fig. 7).
DISCUSSION
As reported previously [1,30], we found that fenbendazole arrests cell cycle at G2/M phase and induces apoptosis via p53-p21 pathways in SNU-C5 and SNU-C5/5-FUR cells. Cell death pathways were further investigated in terms of apoptosis, autophagy, ferroptosis, and necroptosis to identify the differences in 5-FU resistance. Benzimidazoles induce apoptosis via caspase-3-PARP activation [1], which is also true in SNU-C5 and SNU-C5/5-FUR cells with the increased levels of mitochondrial injury marker, cytochrome-C. Benzimidazole is historically known to bind beta-tubulin, disrupt microtubules, and arrest cell division [1,30,33-35]. Although Beclin-1 was increased in SNU-C5 and SNU-C5/5-FUR cells, autophagy might be not so effective due to insufficient LC3 (II/I) activation especially in SNU-C5/5-FUR cells.
Benzimidazole-induced ferroptosis has yet to be fully reported. Ferroptosis is characterized by lipid peroxidation. It can be induced by the accumulation of free iron and via the inactivation of cysteine uptake by SLC7A11 or the inactivation of the lipid repair enzyme GPX4 [36-38]. Free iron level might be regulated (decreased) by the increased expression of FTH1 (storage) and FPN (exporter), whereas decreased expression of GPX4 and/or SLC7A11 induces ferroptosis in SNU-C5 and SNU-C5/5-FUR cells. The effects of anti-ferroptotic agents were elucidated to establish ferroptosis as main cell death pathway in fenbendazole treatment. Treatment with ferrostatin-1 (the inhibitor of SLC7A11-dependent ferroptosis) and DFOM (the iron chelating agent to inhibit ferroptosis) did not effectively block the fenbendazole-induced cell viability, suggesting that ferroptosis is not the primary cell death pathway in fenbendazole-induced anti-cancer effects. However, ferroptosis-inducing agents trigger synergistic apoptosis by increasing DAMP as reported in HCT116 cells [39].
Benzimidazole-related necroptosis has yet to be investigated comprehensively. Necroptosis is negatively regulated by caspases and initiated by a complex containing of RIP and RIP3 kinases, leading to phosphorylation of MLKL [40-42]. In this study, MLKL activation was not affected by fenbendazole treatment, and thus necroptosis was not effectively activated in both CRC cells. But active and intermediate form of caspase-8 was increased in SNU-C5 and SNU-C5/5-FUR cells, respectively, which induces apoptosis via cytochrome-C or caspase-3 [43].
During MAPK signaling, benzimidazole usually inhibits ERK activation [7,44], but increases p38 [5,35] and JNK activation [5,23,33,35,45]. Interestingly, ERK-dependent activation of heat shock factor-1 (HSF-1) promotes chemotherapeutic resistance to benzimidazole (parbendazole and nocodazole) in HCT116 and RKO cells. Biochemical inhibition of HSF-1 results in significant enhancement of drug potency. Therefore, Wales et al. [34] suggested that targeting ERK signaling pathway is a potential strategy in benzimidazole-resistant CRC cells. Despite the contrasting ERK response to yeast extract treatment in SNU-C5 and SNU-C5/5-FUR cells [19], we found no significant changes on p38, ERK and JNK, signaling after fenbendazole treatment.
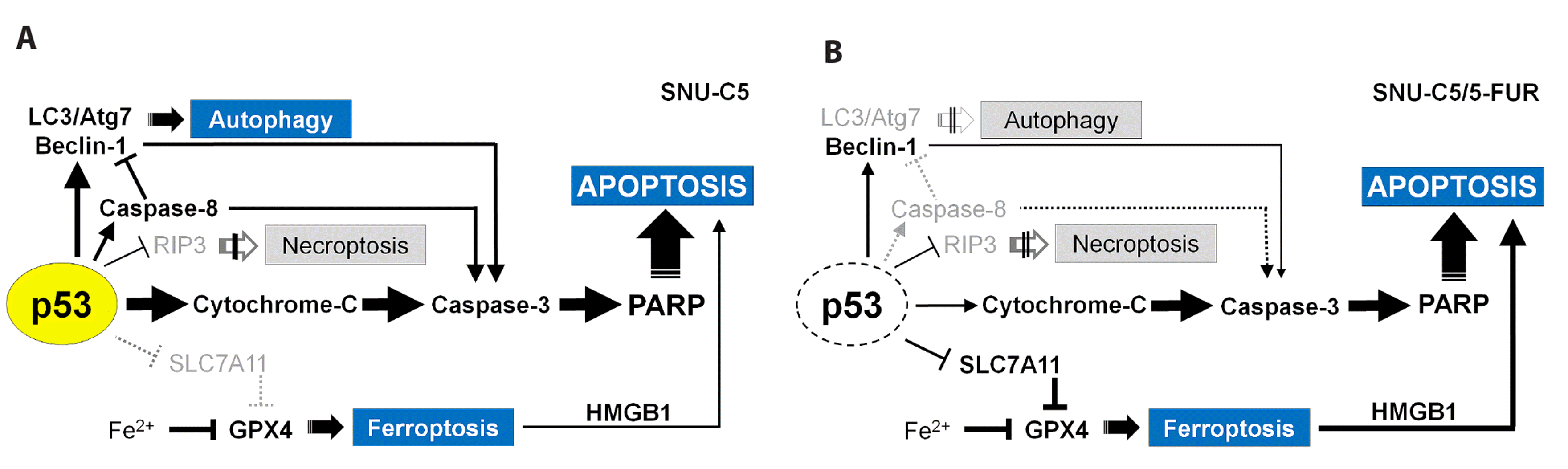
Benzimidazole is known to activate p53 and p21 but decrease mutant p53 expression [30,46]. Cancer cells with wild-type p53 showed higher sensitivity to fenbendazole compared with p53 mutant cells [5,35]. In addition to cell cycle arrest via p53-p21 pathways [1,30,47], p53 acts as a key metabolic switch in cellular function and death [48-50]. In this study, fenbendazole induces G2/M arrest and apoptosis in both 5-FU-sensitive SNU-C5 and SNU-C5/5-FUR CRC cells. In SNU-C5 cells, fenbendazole is presumed to activate p53-mediated apoptosis by increasing p53 expression, and partly necrosis, autophagy and ferroptosis (Fig. 8A). P53 activation is mainly associated with mitochondrial injury and caspase-3-dependent apoptosis, of which caspase-3 activation might be partly induced by caspase-8 and Beclin-1. In addition, ferroptosis by decreased GPX4 might augment apoptosis as previously described [39]. In SNU-C5/5-FUR cells, fenbendazole triggers apoptosis without affecting p53 expression (Fig. 8B), which was characterized by the following differences as compared with SNU-C5 cells. The expression of LC3, Atg7, and active caspase-8 was not increased, which resulted in reduced autophagy and caspase-8-dependent apoptosis. The expression of SLC7A11 as well as GPX4 was suppressed, which resulted in ferroptosis as previously reported [51,52]. As a result, apoptosis was partly induced by Beclin-1, and further augmented by ferroptosis in SNU-C5/5-FUR cells than in SNU-C5 cells. However, the activation of p53 with benzimidazole treatment is still disputed. Benzimidazole induces concurrent apoptosis and pyroptosis of human glioblastoma cells as well as inhibits cell cycle via p53-p21 pathway [53], while mebendazole was associated with p53-independent induction of p21 and tubule depolymerization in ovarian cancer [54]. Therefore, cell-type specific activation of p53 should be identified prior to benzimidazole, including fenbendazole, treatment of various cancers.
Taken together, SNU-C5/5-FUR cells exhibited approximately 10-fold higher IC50 values with fenbendazole compared with SNU-C5 cells, but still showed cytotoxicity at micromolar concentrations as previously reported [35]. Fenbendazole induces apoptosis as well as cell cycle arrest at G2/M phase via p53-p21 pathways in CRC cells. In case of cell death, fenbendazole induces primarily apoptotic cell death rather than autophagy, ferroptosis, and necroptosis. Although fenbendazole has anti-cancer effects on both 5-FU-sensitive and resistant CRC cells, the mechanism of action appears to be different. That is, fenbendazole promotes cell death by activating p53-mediated apoptosis in SNU-C5 cells, whereas by both enhancing p53-independent apoptosis and ferroptosis-augmented apoptosis in SNU-C5/5-FUR cells. Therefore, further studies are needed to identify sensitive and resistant cancer cell types to facilitate cell type-specific treatment with benzimidazole.







 XML Download
XML Download