

 PDF
PDF Citation
Citation Print
Print



INTRODUCTION
Quantitative laboratory measurements are performed by establishing the relationship between the observed instrument signal and the measurand concentration. This relationship is most commonly established using an assay-calibration procedure. Calibration involves testing a set of standards with known analyte concentrations to obtain an instrumental signal response. This relationship is mathematically defined by regression modeling of the measured signal and analyte concentration [1]. Subsequently, a sample of unknown analyte concentration is subjected to the same measurement procedure, and the generated signal is used in the regression equation to interpolate the analyte concentration in the unknown sample.
Calibration is a critical component of the reliability, accuracy, and precision of laboratory measurements. The quality of quantitative data is highly dependent on the quality of the fitted calibration. A poorly calibrated instrument may show a clinically unacceptable bias, leading to negative patient outcomes. Similarly, highly variable calibration affects the precision of the reported results.
Clinical mass spectrometry measurement procedures are generally quantitative in nature and rely heavily on fitted calibration models. It is important to understand the underlying principles of calibration processes as well as the advantages and disadvantages of different regression approaches to ensure optimal practice for a clinical mass spectrometry laboratory.
Commercial and governmental guidelines vary with regard to the requirements for calibration procedures, such as the number of calibrator points and replicates, working calibrator range, and calibrant spacing [2]. Zabell, et al. [2] summarized the suggested practices and highlighted differences in three guidelines with relevance to clinical and preclinical markets: the European Medicines Agency, Eurachem, and United States Food and Drug Administration (USFDA). Regulatory authorities may also propose guidelines without necessarily providing an explanation or evidence for their recommendations. For example, the USFDA requires the use of a minimum of six non-zero calibrators and a zero standard but does provide reasoning as to why at least seven points are needed in the construction of a calibration curve [2].
Although considerable resources are invested in most aspects of full method validation, the rationale behind calibration curve construction, evaluation, and implementation is often overlooked. Common misunderstandings made when considering calibration procedures include the use of correlation coefficients (r) or determination coefficients (R2) to assess linearity and unrecognized heteroscedasticity in calibration data, leading to improper selection of weighting factors [3]. Application of an inappropriate calibration regression model can be a potential source of bias and imprecision in the measurements.
This systematic review was undertaken to examine how calibration practices are applied to liquid chromatography coupled with tandem mass spectrometry (LC-MS/MS) methods used in clinical mass spectrometry laboratories and to provide general guidance for the establishment of optimal calibration practices.
Commercial calibrators may have manufacturer-suggested calibration practices that should be judiciously modified only with sufficient data and expertise to demonstrate their impact on improving analytical performance. However, the post-analytical calibration practices summarized in this review could still be applicable with the use of inhouse-prepared or commercial calibrators.
Go to : 
LITERATURE REVIEW STRATEGY
The systematic literature review was conducted by searching the electronic database PubMed from the date of database inception to April 1, 2022. The search terms used were “calibration,” “mass spectrometry,” and “regression.” Studies were included if they examined regression approaches for calibration in clinical mass spectrometry applications and were published in English. Studies were excluded if they reported on method development without a specific examination of regression approaches for calibration in clinical mass spectrometry applications.
The titles of all retrieved articles were reviewed to exclude non-English and non-relevant studies. The abstracts were then reviewed to select relevant articles for full-text reading. The bibliographies of the selected articles after full-text reading were reviewed to identify other relevant references. The review of all articles was assessed independently by two co-authors (WLC and TPL), and differences in assessments were resolved through discussion.
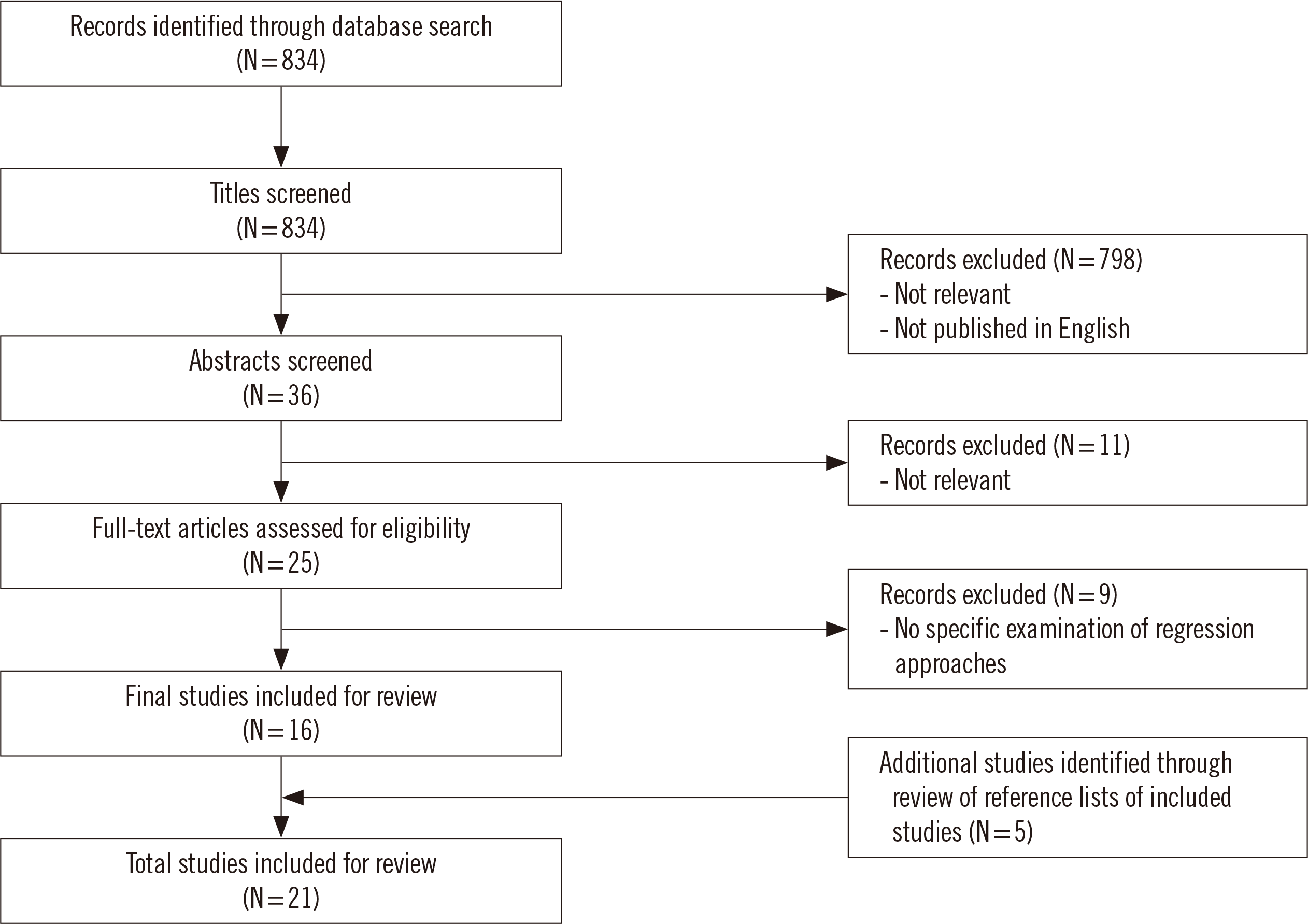
The database search identified 834 publications. These titles and abstracts were evaluated for relevance, with 798 articles excluded through title review, and a further 11 articles were excluded through abstract review. Twenty-five articles were selected for the full-text review, and 16 articles met the inclusion criteria. An additional five relevant articles were identified from the reference lists of the included articles to provide a total of 21 studies included in this systematic review (Fig. 1).
Go to :
DEFINITIONS OF LINEARITY
The term linearity has several meanings because there are different ways to describe linear functions. Linearity refers to the response function term used to describe the relationship between the instrumental signal response and the concentration (calibration function). Linearity also refers to the relationship between the quantity introduced (input) and the quantity back-calculated from the calibration curve (output) [3]. Linearity also has a graphical and mathematical meaning as a linear (straight-line) as opposed to a non-linear (quadratic) regression model used to describe the calibration curve. The scope of this review was restricted to the linearity of the instrument response function, calibration curve, and regression models.
Go to :
RECOMMENDATIONS
Calibration materials, matrices, and internal standards
Calibration matrix
A key assumption in the calibration process is that the signal-to-concentration relationship is fully conserved between the calibration material matrix and the clinical sample matrix. A long-standing recommendation is that calibrator standards should ideally be prepared in matrix-matched materials to avoid bias resulting from matrix differences between patient samples and calibrators. However, the effectiveness of a matrix-matched calibration approach is related to the commutability of the calibration matrix and how representative it is of clinical patient samples [4]. If there are significant matrix differences between calibrators and clinical patient samples, the signal-to-concentration relationship may not be conserved, leading to biased measurements.
For the measurement of exogenous analytes, blank matrices (i.e., matrices devoid of target analytes) from commercial sources or inhouse preparations are readily available. The measurement of endogenous analytes poses a greater challenge, as a “proxy” blank matrix is required for the preparation of calibrators. These matrices (commercial or inhouse-prepared) are often generated through the removal of analytes by dialysis, stripping the native matrix with activated charcoal, or using synthetic matrix materials [5]. Subjecting the matrix to more rounds of stripping (e.g., triple-stripped serum) may reduce the quantity of endogenous analytes in the matrix. However, these additional processes may cause the blank matrix to deviate from the native human matrix and become less representative of the clinical patient samples.
Other variables to consider for calibrator matrix preparation are when the calibration matrix cannot be depleted (e.g., in the case of amino acids), when there is qualitative or quantitative difference in binding proteins of stripped matrix, compared to the ones in native human matrix [6], and in the matrix preparation of unstable molecules. These cases may require spiking of additional components into the blank matrix, such as bovine serum albumin—for nonspecific binding, and antioxidants or preservatives for stabilizing unstable analytes. A synthetic matrix or solvent-based calibrator may be considered when endogenous analytes cannot be effectively removed [5].
It may be desirable to verify the commutability of the calibrator matrix during method development, which can be performed following the CLSI EP07 guideline [7]. Evaluation of differences between the calibrator matrix and native patient matrix, such as spike and recovery experiments, can be conducted to determine the presence of matrix effects [5, 7, 8].
A matrix effect may enhance or suppress ionization of the analyte or internal standard (IS), leading to over- or underestimation of the analyte concentration, respectively. The extent of matrix effect interference can be variable and unpredictable; it may be dependent on interactions between the target and co-eluting molecules or may have a nonspecific effect on instrument responses. The same analyte can give different responses in different matrices, and the same matrix can affect various analytes differently [4, 9]. Matrix effects can be assessed by examining the recovery of a spiked analyte in the matrix under investigation or by observing signal enhancement or suppression by co-infusing a blank matrix with a pure labeled/unlabeled standard [10, 11]. These effects can be reduced using selective sample extraction steps, diverting flow to waste to reduce ion source contamination, or adopting matrix-matched calibration strategies; however, these practices may not completely negate all matrix effects [4]. The matrix effect may be better mitigated by chromatographically optimizing the resolution between regions of suppression/enhancement and the analyte of interest during method development to separate the analyte from the bulk waste of unretained species.
Internal standards
Use of a stable isotope-labeled (SIL)-IS helps to minimize the issues caused by matrix ion suppression or enhancement during quantitation [9]. An SIL-IS allows for accurate quantitation without the need for matrix-matched calibrators by compensating for matrix effects and any potential losses in recovery during cleanup or extraction processes [9, 12]. Although the absolute response can vary, the response ratio (or relative response) of the analyte to SIL-IS remains the same [13]. As described above, other matrices should be evaluated to determine the effects of matrix differences.
Ideally, the SIL-IS should exactly mimic the target analyte(s) to correct matrix-related responses. An SIL-IS must behave in the same way as the target analyte in both the sample extraction and ionization processes. For this compensation to be effective, it is necessary for the IS to resemble the analyte in terms of physical and chemical properties. The coincidental retention time of two structurally unrelated molecules is insufficient to ensure ideal behavior as an IS. The overall variability of experimental results increases as the structures of the target analyte and IS become more divergent [14]. An IS must be structurally similar and chromatographically co-eluted with the target analyte to effectively compensate for non-proportional ionization due to the matrix effect [9, 15].
Although several options for SIL-IS are available, 13C- or 15N-labeled compounds are preferred over deuterium (2H)-labeled compounds because they demonstrate better labeling stability and often have greater purities. They also display identical or almost identical chromatographic and ionization behaviors to those of unlabeled target analytes [16]. SIL-IS is expected to reduce the effects of matrix ion suppression or enhancement, provided they co-elute from the column. For an SIL-IS labeled with deuterium, the position of deuterium isotopes may lead to hydrogen-deuterium exchange, causing IS response variability, leading to the loss of deuterium isotopes in some cases. An SIL-IS heavily labeled with deuterium isotopes may not co-elute with the target analyte because of slight differences in physiochemical properties, resulting in partial ionization differences in the matrix [17, 18]. Importantly, a certificate of analysis for an SIL-IS usually indicates purity as a function of liquid chromatography-UV analysis, which is not capable of indicating isotopic purity.
Signal drifts can be observed during an analytical run, which may be caused by fluctuations in liquid chromatography hardware performance, variations in the electrospray process, changes in ion transfer caused by fouled or moved optics, and changes in detector sensitivity [9, 19]. Among these variations, electrospray ionization at the ion source is considered the major cause of instrumental response fluctuations. An IS is commonly used in quantitative analyses to compensate for these drifts using the analyte-to-IS ratio [19, 20].
Number of calibrator standards, analysis, and positioning of calibrators
Constructing a calibration curve
A minimum of two data points is needed to draw a straight-line graph and for linear regression modeling (first-degree polynomial) according to Equation 1:
where y is the instrument response (or normalized ratio), x is the concentration of the analyte, a is the analytical sensitivity of the instrument (i.e., the signal per unit change in the concentration of an analyte [21], or slope), and b is the instrument response when no analyte is present (or intercept).
Increasing the polynomial order of the equation to a second-degree polynomial would require a minimum of three data points to plot the curve, and a further increase to a third-degree polynomial requires four data points. In general, a higher number of data points is required for non-linear regression. Non-linearity in calibration curves is commonly observed in LC-MS. Common causes of the observed non-linearity are matrix effects, saturation during ionization, dimer or multimer formation, isotopic effects, and detector saturation [19, 22]. If known causes of non-linearity can be mitigated during method development, they should be implemented to achieve a linear calibrator response. However, if non-linearity remains at the end of method development, non-linear curve fitting may be considered, as elaborated below.
Number of calibration levels
The aim of regression modeling calibration data is to minimize the error in the estimation of parameters describing the relationship between the standard concentration and observed signal response. Increasing the number of points in the calibration curve reduces the uncertainty in the estimated parameters. A sufficient number of calibration standards is needed to define the response profile in relation to the concentration range of the standards. There is no agreement on calibration strategies from regulatory bodies and organizations regarding the number and concentrations of calibrators required, which are often arbitrarily selected. One common recommendation by commercial guidelines and regulatory authorities is that a calibration curve should include a minimum of six non-zero samples covering the intended calibration range, a zero sample (matrix with only the IS), and a blank sample (matrix without any analyte or IS) [2, 23]. The non-zero calibrators should also encompass the lower limit of quantitation (LLOQ) and upper limit of quantitation (ULOQ) [2, 23]. Zero and blank samples (also known as blank and double blank, respectively) should not be included in the calibration curve regression [5]. They are to be utilized to ensure that the veracity of the system does not unduly bias the intercept.
Having a greater number of calibration standards leads to a smaller error estimate for the calculated concentration and, consequently, better precision and a narrower confidence interval at the determined concentrations. Although more calibration points can improve the accuracy and precision of the model, this must be balanced against operational and financial costs such as greater laboratory effort in the preparation and analysis of additional calibration standards or replicates [24].
Similar accuracy and precision can be achieved by using as few as one calibration standard (with the regression being forced through the origin) or two calibration standards (without the regression being forced through the origin) to construct the calibration curve as against using eight or ten standards [25-27]. Under the two-calibration standard approach, previously established linear multipoint calibration data were retrospectively linearly regressed using two critical concentrations, the LLOQ and ULOQ, with no additional batch failure or QC rejections observed. More specifically, the differences in mean QC concentrations were within −0.8% to 2.5%, and the differences in %CV were within −0.7% to 0.9% for the 12 measurement procedures studied [27]. These studies involved exogenous analyte applications, which may be associated with better opportunities for matrix matching between the calibrators and samples. It is important to determine the susceptibility of a low-number calibration strategy to calibration drift or shift errors, and its ruggedness over time.
An increased number of points in a calibration curve is only an indication of the back-calculated values of the standard curve points and is not directly correlated with the inaccuracy or imprecision of points that were not used to generate the model (e.g., QCs or samples). Additional calibration points/replicates may improve the precision of the regression model, but this does not directly correlate with improved performance of the assay. This may be especially true for assays with modified matrices (e.g., endogenous analytes using a stripped matrix).
Tan, et al. [26] demonstrated that the impact of using different high-concentration levels as calibration points was almost identical. Hence, the ULOQ calibrator can be represented by adjacent high-concentration standards close to the ULOQ, and extrapolation at the higher concentration end is generally linear and acceptable [26]. The addition of a concentration point at the geometric mean of the LLOQ and ULOQ can establish quadratic regression models. Placing a calibration point at the geometric mean also provides better overall accuracy than using other adjacent points, although extrapolation beyond the highest concentration is not advised [26]. These results should be interpreted within the specific simulation parameters. For example, the degree of quadraticity of the regression curve may vary from day to day depending on the condition of the source/ion optics/detector. The application of such a strategy in other LC-MS/MS techniques warrants examination.
It may be difficult to determine the true linearity of a calibration curve using only two points. Musuku, et al. [27] demonstrated alternative approaches to verify the linearity of the curve, such as performing a regression using the two calibrators together with QC samples; however, this may not always be sufficient to conclude linearity. It is advisable to validate the method using more calibrator concentrations first to map out the detector response and investigate polynomial regression models, while conducting experiments to critically stress the linearity assumption before subsequently removing some calibration levels to maintain the performance specifications [2, 26]. It may be necessary to remap the detector response when a significant analytical shift or drift is observed.
Number of replicates
The above studies show that optimal accuracy for regression statistics can be obtained using fewer calibration concentrations with a higher number of replicates at each concentration, instead of the commonly adopted practice of using more calibration concentrations with fewer replicates [26]. Additional replicates help to reduce the imprecision of the estimated parameters [24]. Having fewer calibration standards also reduces the risk of error from calibration standard preparation. The number of replicate calibrator injections is also an arbitrarily set criterion. Duplicate injection of calibrators is recommended because it improves the precision of the observed result without requiring additional calibration standard preparations [23, 24, 26].
The number of calibrator points and replicates analyzed, as well as their concentration levels, should be dependent on the characteristics of the particular bioanalytical measurement procedure, such as the presence of linearity or non-linearity and the precision of the analysis [26].
Order of calibrator assessment
The order of calibrator assessment does not significantly influence the performance and can be conducted in either ascending or descending order [23]. Nonetheless, the addition of a blank sample after injection of the highest-concentration calibrator can assist in the detection of carryover [23].
Having the calibrations bracket an analytical run, with one set at the beginning and one at the end of a sequence, can help reduce the effect of analytical drift throughout the run. Combining both sets of data points to construct a calibration curve helps to compensate for any variations in instrument sensitivity (slope changes) during the run. If only one replicate injection of the calibration is adopted, interspersing the calibrators in the run can achieve the same compensation [24].
Distribution of standard concentrations
Another related issue is how calibration levels should be distributed across the measurement range. The concentration levels can be evenly (equidistant) or unevenly distributed. Unequal spacing of calibration points with clustering at lower concentrations improves the accuracy and stability of the calibration curve at the lower end, which subsequently improves the precision of measurements near the limit of detection and LLOQ. Having a single very high calibrator concentration (i.e., spaced far away from other points on the x-axis) may lead to a high degree of leverage, with a small error having a disproportionately large influence on the regression model estimates [2].
Although not thoroughly examined in the literature covered for this review, different distributions of calibrators can influence stability of the regression model when an incorrect weighting factor is used. With an appropriate weighting factor, the curve was stable regardless of the calibrator distribution [28]. Weighting the response also reduces leverage effects [2]. Further research is required in this area of calibration data heteroscedasticity, precision profiles, and appropriate regression model weighting.
It is generally considered suboptimal to prepare calibrators by serial dilution from a single stock because this carries a higher risk of error if the primary stock is prepared incorrectly.
Frequency of calibrations
Another area of interest in determining calibration practices is how often a calibration procedure should be performed. Conventionally, calibrations are performed for each sample analysis batch.
Alladio, et al. [16] demonstrated the possibility of collecting calibration data from analyses over a few weeks to build a robust averaged calibration curve using gas chromatography-mass spectrometry. It may be advantageous to use this averaged calibration curve built from data collected on different days, giving a larger number of replicates, instead of changing the calibration curve daily, especially when the instrumental conditions are stable. The authors cautioned that this conclusion might not hold for LC-MS/MS methods because of the larger between-day variation [16].
Early evidence suggests that this averaged calibration approach may indeed be useful for LC-MS/MS analysis. In one study, tacrolimus values extracted together with calibrators were comparable to the values extracted within 24 hours of the initial calibration [29]. This would be an interesting area for further study, as a reduction in calibration frequency could result in significant operational and financial savings.
Calibration curves were constructed to associate the analytical response with the standard concentration, and QC was used to confirm the response associated with the concentration and monitor the performance of the measurement procedure. The robustness of a measurement procedure is defined by the stability of its performance [2].
Other factors may influence the calibration stability. The SIL-IS is a global normalization factor that relates to the historic calibration curve. Any environmental or analytical change that dissociates the SIL-IS from the historic calibration curve and contemporaneous samples may introduce analytical errors. Variations across analytical batches arising from reagent preparation, instrument conditions, and technical expertise may cause shifts in the calibration curve [1]. Calibration with each batch to recharacterize the instrument does not resolve the actual variability but could be an inappropriate soft correction for possible day-to-day variances, while presuming that the calibration is error-free [2]. Zabell, et al. [2] placed a stronger emphasis on using QC performance to indicate recalibration or after major changes in reagents or instrument conditions.
Calibration modeling (regression approaches and weightage)
When examining calibration data for fit, it is important to consider the regression method (e.g., least-squares, Deming), model (e.g., linear or polynomial), and fitting technique (e.g., weighting). Selecting an inappropriate regression approach for a calibration procedure could lead to significant bias and imprecision by modeling an inappropriate relationship between the signal measurements and standard concentrations. The selection of a correct regression model during the method development and validation stages is critical for a smooth transfer from the method validation stage to production, which should also be maintained in the production stage [28].
Linear or non-linear regression models
As mentioned above, it is common to observe non-linear calibration data using LC-MS methods. Non-linear behavior may not be evident from visual inspection of the calibration data but may display significant bias when fitted with an inappropriate curve. When non-linearity is determined in calibration data, there are two options to overcome this limitation. First, a quadratic or higher-order calibration regression model can be used. Second, the calibration data can be divided into two separate ranges. A linear regression model may be used to fit the lower calibration ranges. This lowers the calibration range to narrower linear ranges, reducing the dynamic range of the measurement procedure [14, 15, 30].
Although r and R2 are commonly used indicators to assess the goodness of fit for calibration models, they are not appropriate for assessing linearity [31]. Both r and R2 are statistical measures; r is an indicator of the degree of correlation between two variables (signal and concentration), and R2 is an indicator of the proportion of variability in the response explained by the regression. The correlation and response variability are only loosely related to linearity, and using these two coefficients to determine linearity may be misleading. These coefficients used in isolation are not adequate to assess linearity because values close to unity (e.g., R2 >0.999) can be obtained even when the data show signs of curvature [3, 32]. Linearity should instead be assessed using appropriate statistical methods (e.g., ANOVA) and/or other mathematical measures (e.g., residual plot), which will be further discussed in Section 5 below.
The two most commonly used regression models for constructing LC-MS/MS calibration curves are linear and quadratic regression equations, which use either weighted or non-weighted fitting techniques [1]. To determine whether the calibration model should be weighted, the calibration data should be examined for evidence of heteroscedasticity.
Regression fitting technique
Heteroscedasticity in regression analysis refers to the error term or residuals, which are unequal across the values of the dependent variable (calibration standards in this case). Calibration data may be homoscedastic, where the variance of each concentration level is constant and independent of the concentration range, or heteroscedastic, where the variance increases as a proportion of the concentration range. When calibration data are heteroscedastic, a scatter plot of these variances often shows a funnel shape, in which the variance widens or narrows in response to the standard concentration [32]. A standard non-weighted (ordinary least-squares) calibration regression model assumes that the measurement error is homoscedastic (i.e., exhibits constant variance). If calibration data are heteroscedastic, it is more appropriate to use weighted least-squares regression [1].
When the calibration data are heteroscedastic, but no weighting is applied during regression modeling, the influence of errors at different concentration levels on the estimation of the parameters is ignored, which reduces the stability of the calibration models. Model stability is defined as the resistance of a calibration to significant errors from calibration samples [28]. Using a non-weighted regression means that a small bias at higher calibration concentrations could change the curve significantly and cause large deviations at low concentrations. This is especially detrimental for heteroscedastic data because the variance at each level could increase with increasing concentrations.
To account for the heteroscedasticity of the data, weighted regression analysis has been used to maintain constant variance through the measured concentration range. Weighted regression models minimize the influence of higher concentrations by balancing the regression line to distribute the variance uniformly throughout the calibration range [31]. Appropriate weighting factors can be calculated using the inverse of variance (1/σ2). However, this practice requires several determinations for each calibration point [31]. Instead, weighting methods commonly involve adjusting the data using a factor related to an inverse function of the concentration of the standards. Weights of 1/x2 have been recommended as LC-MS/MS bioanalytical methods [26, 28, 33]. Simply following historical practices may not be appropriate, as many of these recommendations are based on the “test-and-fit” strategy, which involves fitting the calibration data points with different models and weighting factors, starting with the most simplistic unweighted linear regression and progressively fitting more complex models and subsequently assessing which model provides the best fit. This is often largely based on the analyst’s subjective interpretation that the data points are close to the trend line and that the regression has an R2 value close to unity.
Good recovery of the calibration standard concentrations and/or QC performance does not necessarily mean that a correct weighting factor has been applied. Acceptable recovery may be achieved when an inappropriately weighted calibration model coincidentally overlaps or is close to the underlying true relationship. Gu, et al. [28] demonstrated that, in some cases, no weighting or 1/x weighting could generate good calibration curves and QC performance, although the weighting factor determined from the collected data should have been 1/x2. Hence, recovery and assay performance data should not be used as criteria for the selection of weighting factors. The choice of weighting depends on the relationship of the variance for the data with other variables, and correct application of weighting factors generally results in better longitudinal assay performance and stability, as explained further below in Section 5.
Other weighting factors that are less commonly used in clinical LC-MS/MS applications are 1/y and 1/y2. In most cases, the effects of using either 1/x or 1/y or 1/x2 and 1/y2 are similar for bioanalytical LC-MS/MS measurement procedures, as linear or quadratic models with very mild curvatures are commonly encountered. Instrument response errors (or variances) are directly related to the y-axis instead of the x-axis on the calibration curve plot. When the curves are linear or close to linear, the empirical functions between 1/σ2 and y can be translated into the same empirical functions between 1/σ2 and x [28].
When the quadratic curve has strong curvature, or for the four-parameter logistic and five-parameter logistic curves commonly used in ligand-binding assays, the empirical function between 1/σ2 and y cannot be translated to the same empirical function between 1/σ2 and x. In such cases, 1/y or 1/y2 weighting factors are preferable [28, 34].
Impact of heteroscedasticity on calibration performance
Heteroscedasticity in calibration data should not be overlooked. Heteroscedasticity can lead to a significant loss of precision, particularly at low concentrations, in the calibration model. This is crucial in clinical mass spectrometry as it affects the limits of detection and quantitation of the measurement procedure.
Non-weighted regression modeling of calibration data that are heteroscedastic leads to an increase in imprecision, particularly at lower concentrations, resulting in falsely higher limits of detection and quantitation and incorrect performance characteristics of the measurement procedure. Alternatively, application of weighting to homoscedastic calibration data could lead to a falsely lowered calculation of detection and quantitation limits [14, 30-32].
Validation and statistical assessment of calibration models
Several studies recommended simple procedures for selecting the correct regression model and weighting factors for calibration models based on the experimental data collected [14, 28, 32]. Using a combination of graphical plots with visual assessment and statistical methods, the calibration linearity and homogeneity of variances can be evaluated. It is highly recommended to use these methods to determine the correct regresregression approaches for the measurement procedure.
Several procedures can be followed to test whether the experimental calibration data are homoscedastic or heteroscedastic and whether a weighting factor should be applied. This can be performed graphically, visually (qualitatively), or through statistical approaches (quantitatively).
For graphical methods, a scatter plot of the residuals derived from unweighted least-squares regression versus the predicted values can be generated [31, 32, 35]. The residual (R), which is the difference between the measured values (Sexp) and the calculated or fitted values from the regression equation (Sint), can be established using Equation 2 [31].
where Sexp is the experimental/observed signal and Sint is the interpolated signal derived from the regression equation. The residual is calculated for each calibration data point, and the residuals are plotted against concentration. The graphs are then visually assessed to determine whether the residuals are randomly distributed across the concentration axis (x-axis). A funnel-shaped trend, where the variance is more pronounced at increasing concentrations, indicates heteroscedastic data and that a weighting factor should be applied [31, 32].
Residual plots can be generated using different calibration models (linear or quadratic) and weighting factors. The plot displaying the most symmetrical distribution of the residuals around the concentration axis indicates that the assumptions of the model and subsequent error terms are correct [35]. However, residual plots may not always be easy to interpret, particularly when the number of calibration points is limited.
The percent relative error (%RE) can also be used as a quality indicator in optimal model selection [30]. The %RE can be derived from regression models, as shown in Equation 3, with the deviations from the calibration model determined by comparing the back-calculated concentrations with the theoretical or nominal values of the calibration standards.
In Equation 3, Cexp is the experimental or observed value and Cnom is the nominal or theoretical concentration. The sum of the absolute %RE values is used to determine if appropriate model fitting is achieved for all calibration points. The optimal regression model provides a narrow horizontal band of randomly distributed %RE across the concentration axis and the least absolute sum %RE [30].
The F-test is a statistical approach that can be used to determine if the variances of signals at the lowest and highest calibrator levels differ significantly [14, 32]. If the calibration data are heteroscedastic (at a significance level of P<0.05), a weighted model is a more appropriate choice. Generally, a 1/x weighting factor is used when the variance increases proportionally with the standard concentration, and a 1/x2 weighting factor is used when there is a quadratic increase in variance. The weighting factor that generates the smallest variance of the weighted normalized variances is selected as the optimal factor [14]. Similarly, Bartlett’s or Levene’s tests can be performed to simultaneously compare variances at all concentration levels [14, 32]. An additional benefit of Bartlett’s test is that it can be used to compare variances with unequal sample sizes, which may occur when calibration replicates are excluded from variance evaluation due to poor injections or gross errors [36].
The number of terms for the calibration model can be established by comparing the variances of the linear and quadratic models using a partial F-test [14, 33]. If the quadratic calibration model significantly improves the modeled variance of the data in comparison with the linear model (at a significance of P<0.05), then the quadratic model should be selected. The ANOVA-lack of fit (LoF) test can also be performed to verify the goodness of the calculated calibration model, although Alladio, et al. [14] caution that this test is sensitive to the number of replicates and calibration levels [33].
Statistical tests or mathematical functions, in comparison to graphical assessment, are less empirical approaches for determining linearity. However, any issues occurring in the measurement procedures, such as nonspecific adsorption, cross-contamination, systematic bias, errors due to preparation or storage, and matrix interferences, may influence the regression models and estimated parameters. Care must be taken to ensure that the data collected and used for statistical modeling are a true and accurate representation of the calibration procedure. Validation of the calibration regression should be conducted over many runs performed over a certain time period to capture more sources of variation.
The calibration curve slope and intercept should theoretically be consistent for a validated measurement procedure over the period of method validation and sample analysis, particularly when an SIL-IS is used. In reality, owing to the factors mentioned above, variances across batches may lead to variability in the slope and intercept. This consistency of calibration curve slope and intercept is often used to assess the robustness of a method [23]. The calibration data collected for validation of the regression model can be monitored for the precision of the curve slopes and/or intercepts to assess the ruggedness of the selected model and reveal any potential issues.
In a regulated clinical laboratory environment, the procedure for determining calibration regression models should be clearly defined in the laboratory’s standard operating procedures. Once the optimal model is determined during method validation, it should not be altered during production. Changing the regression model terms and weighting factor (i.e., changing from a linear to quadratic model or no weights to applying 1/x2 weights) to improve the fit of the calibration model to pass an analytical run should never be undertaken.
Assessment and selection of the calibration model and weighting using only calibration data may run the risk of missing important matrix-related effects in patient samples that are not accounted for by the calibration material. For example, a calibrator using a depleted matrix may behave differently from a patient sample, which may not be fully accounted for by the SIL-IS. Consequently, the signal-to-concentration relationship may not be conserved between calibrators and patient samples, leading to analytical errors. The validation requirement of the calibration curve should consider the clinical utility of the measurement procedure, including the dynamic range and clinical interpretation of the results.
Validation of the calibration model focuses only on the mechanics of calibration practice. Further metrics for validation, such as accuracy, precision, linearity, and determination of the measurement limits, should be adopted. The methods used to validate the calibration regression are summarized in Table 1.
Table 1
Validation and assessment of calibration regression
| Method | Interpretation | Acceptance criteria | Other comments |
|---|---|---|---|
| Plot relationship between residuals and concentration [31, 32, 35] | The optimal regression model and weighting factor will result in randomly distributed variation around the concentration axis. | Not available | Quick and easy graphical visualization; reveals whether the assumptions on the errors and the model are correct. May not always be easy to interpret, especially when the data size is limited. |
| Relative errors and sums of relative errors [30, 31] | The optimal regression model will result in the least absolute sum of relative errors and a narrow distribution band in a plot of residual error against concentration. | Acceptable deviation in relative error is 20% at the lower limit of quantitation and 15% for the rest of nominal concentrations [36]. |
Less empirical approach to assessing the linearity of a calibration curve compared to graphical visualization. Acceptance criteria of 15%–20% could be considered excessively high [3]. |
| ANOVA F-test to compare the variance of the signals at the lowest and highest calibrator concentration levels [14, 31] | Data are heteroscedastic if P < 0.05. | Acceptance limit based on statistical significance. | None |
| Bartlett’s or Levene’s test to compare the variances of replicates at all concentration levels [32, 35] | Test for homogeneity of variances. Data are heteroscedastic if P < 0.05. | Acceptance limit based on statistical significance. | Bartlett’s test can be used to compare variances with unequal sample sizes. |
| ANOVA partial F-test to compare the variances of linear and quadratic models [14] | If the quadratic calibration model significantly improves the captured variance of the data in comparison with the linear model (P < 0.05), the former is accepted. | Acceptance limit based on statistical significance. | None |
| ANOVA-lack of fit test [14] | Lack of fit of the regression model is determined if P < 0.05. | Acceptance limit based on statistical significance. | Sensitive to the number of replicates and calibration levels. |
![]()
Internal calibration
There are many challenges with current calibration practices in clinical mass spectrometry, as discussed above. Internal calibration is a novel approach, which obviates the use of external calibration curves [13, 16, 37-39]. Currently, an SIL-IS is commonly used to normalize instrumental responses and attenuate the overall analytical variation caused by random errors, matrix effects, poor recovery, or instrumental drift [16].
Internal calibration is an alternative approach based on the measurement of isotopic abundances with subsequent isotopic pattern deconvolution. When an SIL-IS is added to the sample, the altered isotopic abundances represent a mixture of a linear combination of those from naturally occurring isotypes and those from the spiked labeled standards. Multiple linear regression is then used to deconvolute the resulting combined distribution of abundances to obtain their molar fractions [37]. The concentration of the measurand in the unknown sample is calculated from the ratio of the signals of the unknown analyte to the SIL-IS multiplied by the SIL-analyte concentration equivalent [16].
This approach overcomes some of the challenges of conventional external calibration curve approaches, such as matrix effects, because the SIL-IS is spiked into patient samples, thus eliminating the difference between the matrices of calibration standards and clinical samples. Internal calibration also reduces the effects of signal drift because the analysis occurs concurrently and can be performed without the need for sample batching with calibrators. Another advantage is a simplified concentration calculation that does not require the plotting of calibration curves and the resulting selection of models, methods, and weighting [9]. Nonetheless, such novel approaches and the required calculations may not be readily available in routine mass spectrometry middleware.
A possible limitation of this approach is that the ULOQ depends on the SIL-IS concentration used. In the studies mentioned above, the SIL-IS concentration was considered to be the upper limit of the studied dynamic range. Additional validation must be performed to determine the linearity of the measurement procedure beyond the SIL-IS concentration. A similar limitation may also apply to the LLOQ, which should be evaluated. The range in which this method provides acceptable measurement performance may be restricted compared with that of traditional external calibration approaches [9].
Other limitations associated with SIL-IS use (see the Internal Standards section above) also affect the internal calibration approach. These include the purity of the SIL-IS labeled with deuterium, hydrogen-deuterium exchange during the course of analysis, and isotope dissociation [40]. The effect of heteroscedasticity on this approach is underexplored and may not be negligible [41].
For widespread use, the internal calibration method needs to be developed diligently to ensure assay linearity and stability of the SIL standards. Experiments must also be performed during validation to examine the effects of the matrix and the time required for SIL standard equilibration with the sample (if any). This approach also requires full characterization of both the analyte and SIL compounds in terms of their isotopic distribution of abundances [37]. Similar to the SIL-IS used for external calibrations, these SILs should resemble the analyte in both physical and chemical properties and must co-elute chromatographically.
Go to :
KEY TAKE HOME MESSAGES AND PRACTICAL RECOMMENDATIONS
A calibration curve is a regression model that estimates the relationship between the known concentration of a measurand and the observed instrument response, which facilitates estimation of the concentration of the measurand in an unknown sample. Calibration plays a critical role in LC-MS/MS analyses; however, insufficient consideration has been given to the decisions behind good calibration practices.
In this review, we briefly summarized the factors to be considered when implementing calibration protocols, such as the importance of using matrix-matched calibrator materials and SIL-IS, the number and concentrations of calibration points, and the non-constant variance in the calibration data.
Although the use of weighted regression results in more complex models than ordinary least-squares regression and requires additional statistical testing, weighting should be considered during method validation to obtain better assay performance specifications, particularly at the LLOQ. The regression method, model, and fitting technique used for the measurement procedure should be tailored to the empirical data-generating process characterized during method validation. Interested readers are encouraged to read two excellent reviews by Rappold [42, 43] to gain additional insights into method development and operationalization of clinical mass spectrometry.
A stepwise approach to determine an optimal calibration strategy for LC-MS/MS bioanalytical measurement procedures is presented below.
Go to :
 XML Download
XML Download