

 PDF
PDF Citation
Citation Print
Print



Dear Editor,
Fanconi anemia (FA) is a rare recessive genetic disorder characterized by genomic instability, congenital malformation, progressive bone marrow failure, and cancer predisposition [1]. At the cellular level, hypersensitivity to DNA inter-strand cross-links is a defining feature of FA [2]. To date, variants in 22 distinct FA genes have been identified to interfere with DNA replication-dependent repair, involving cross-linked DNA at stalled replication forks. Loss of function of related proteins causes hypersensitivity of FA cells to inter-strand cross-linking agents [3]. This pathological phenotype can be ascertained by drug sensitivity assays that expose T lymphocytes to DNA cross-linking agents, such as diepoxybutane (DEB) and mitomycin C (MMC) [4]. Fanconi anemia group D2 protein (FANCD2) western blotting is also used to detect the occurrence of the ubiquitinated isoform of FANCD2 in response to treatment with genotoxic reagents that induce inter-strand DNA cross-links. This assay readily reveals FA in cases in which this isoform is lacking [5, 6]. These functional assays are laborious but nevertheless valid in cases of variants with unclassified significance or those detected in a new putative FA-associated gene. Proof of the pathogenicity of novel variants requires the introduction of the wild-type allele and phenotypic correction. We describe a functional assay to determine molecular and cellular defects using patient-derived dermal fibroblasts from two Korean FA patients with variants in the Fanconi anemia complementation group A (FANCA) gene. Reconstitution of lentiviral transduction of wild-type FANCA successfully complemented the functional defects in the patients’ cells, providing evidence of the pathogenicity of the detected variants.
Written informed consent was obtained from both patients and their parents. The Institutional Review Board of Seoul St. Mary’s Hospital, College of Medicine, The Catholic University of Korea, Seoul, Korea, approved the study (KC11EISI0836).
In February 2010, a 5-year-old boy (patient 1) visited the emergency room with abdominal pain and hematochezia. He also complained of recurrent epistaxis, easy bruising, and pain in the perineal area. The colonoscopic findings were unremarkable, but a blood assay showed thrombocytopenia (78×109/L, reference interval, RI: 150-450×109/L). He presented with short stature, clinodactyly with brachymesophalangia on the bilateral fifth fingers, and multiple café-au-lait spots on physical examination. The DEB and MMC stress assays showed 11.2 and 6.1 chromosomal breaks per aberrant mitosis, respectively (cut-off value: 5.0) [7].
In May 2004, a 9-year-old boy (patient 2) visited a local hospital because of recurrent epistaxis. The complete blood cell count showed 4.3×109/L leukocytes (RI: 4.0-10.0×109/L), 105 g/L Hb (RI: 130-180 g/L), and 39×109/L platelets. Physical examination revealed hypoplasia of the right thumb, short stature, and hypogonadism. His older sister also had polydactyly in her hands. The chromosomal assay revealed a normal male karyotype, and the chromosomal breakage induced by MMC/DEB was within the normal range (0.3/0.75). In April 2006, a chromosomal breakage assay using skin fibroblasts revealed clastogen-induced chromosomal instability.
Genetic diagnosis was made by whole-exome sequencing (Agilent SureSelect Human All Exon V4+UTR kit, Agilent Technologies, Inc., Santa Clara, CA, USA). Both patients harbored two heterozygous variants of FANCA. Allele-specific Sanger sequencing using a 3.8-kb FANCA cDNA fragment encompassing both variants revealed that they were in trans configuration, and the two splice-site variants led to exon skipping and intron retention, respectively. The FANCA mutations in patient 1 were c.2546delC (p.S849Pfs*40) and c.3627-1G>A (r.3627_3642del), and those in patient 2 were c.3091C>T (p.G1031*) and c.426+1G>A (r.426_427ins[ga;426+2_427-1;a>g]) (Fig. 1).
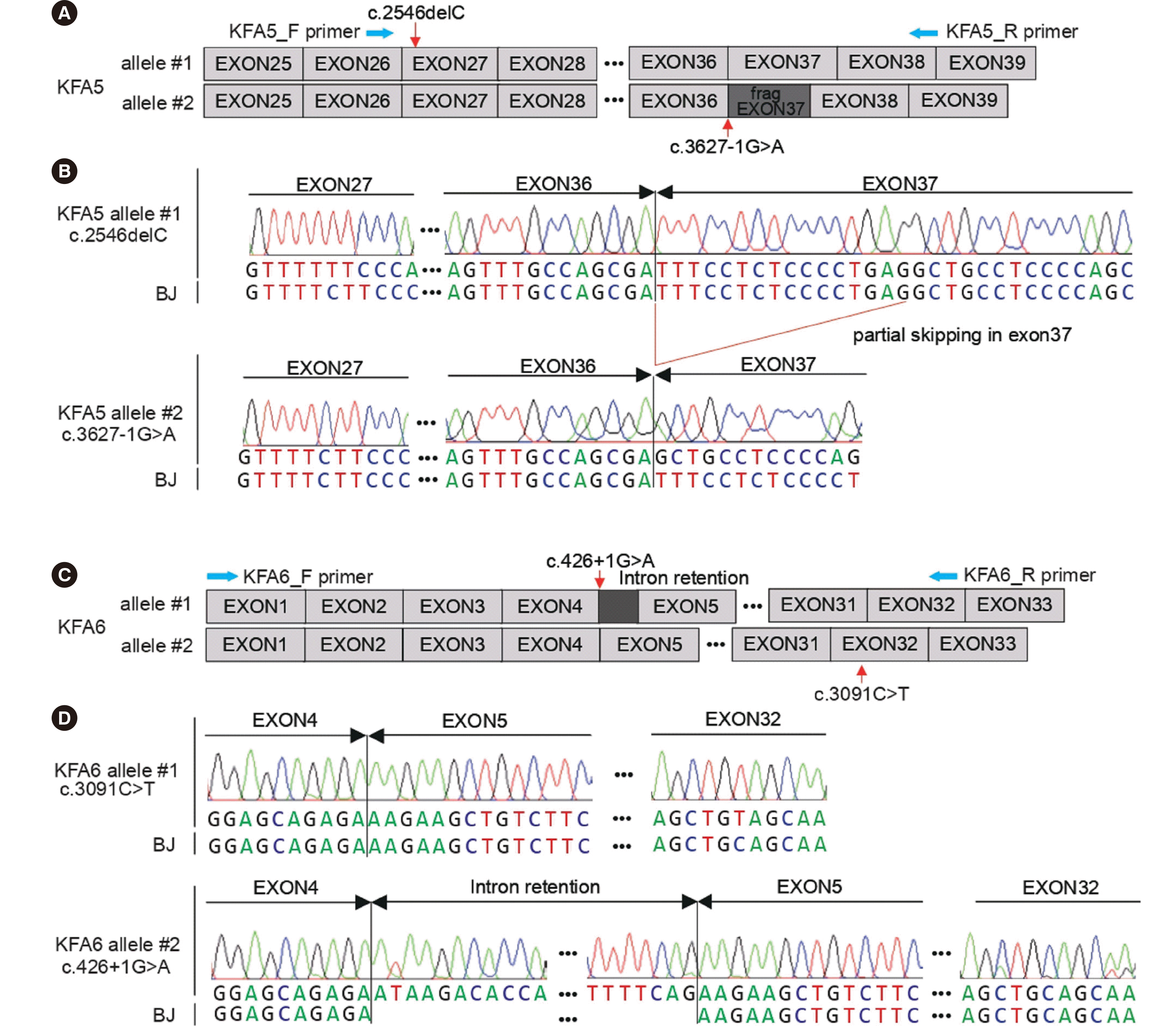 | Fig. 1Allele-specific sequence analysis to identify novel FANCA variants in Korean patients with Fanconi anemia. (A, C) Schematic diagrams of mutant alleles of patient 1 (KFA5) and patient 2 (KFA6). (B, D) Sanger sequencing chromatograms of the patients and normal foreskin fibroblasts (BJ) from American Type Culture Collection, showing that each variant is located in different alleles.
|
Patient-derived dermal fibroblasts were used to examine functional defects due to the detected variants. Western blotting confirmed that endogenous FANCA was undetectable in both cell lines (Fig. 2A). FANCD2 monoubiquitination was not detected in either patient-derived cell line after MMC treatment (Fig. 2B). Patient-derived cells were hypersensitive to MMC compared to the control (Fig. 2C), indicating pathogenicity at the cellular level.
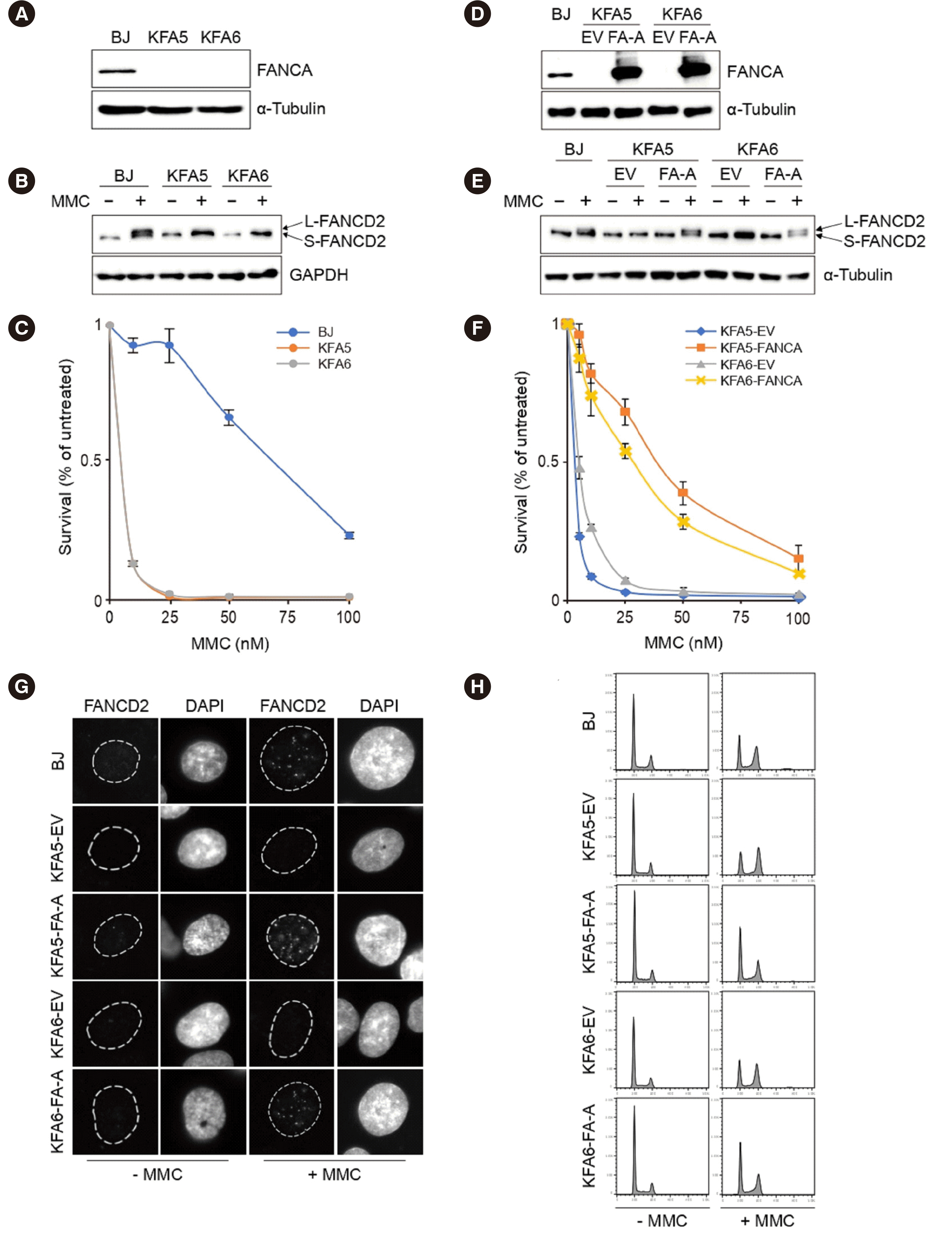 | Fig. 2Molecular and cellular characterization of cell lines and complementation assay. (A) Western blot showing the absence of FANCA protein expression in both cell lines from patient 1 (KFA5) and patient 2 (KFA6), in contrast to the normal fibroblast cell line (BJ). (B) BJ, KFA5, and KFA6 cells were untreated or treated with 1 μM mitomycin C (MMC) for 24 h. Long-form FANCD2 (L-FANCD2) indicates monoubiquitinated FANCD2, and short-form FANCD2 (S-FANCD2) indicates non-ubiquitinated FANCD2. MMC-treated patients’ cells lack the L-FANCD2 bands, representing the FA pathway defect. (C) Patient cells (KFA5, KFA6) are sensitive to MMC treatment compared to the control cells (BJ). Triplicate cells were exposed to four MMC concentrations for 5 days. Percent survival was calculated by dividing the survival rate by that of untreated cells. (D) FANCA protein and L-FANCD2 bands appeared in patients’ cells transfected by FANCA cDNA (FA-A), but not by the empty vector (EV). (E, F) The complemented cell lines shows the successful restoration of function. (G) Immunofluorescence reveals FANCD2 focus formation in the complemented cell lines. (H) Flow cytometry showing cell cycle recovery in the complemented cell lines after MMC treatment.
|
To complement the functional defect, patient cells were transformed by lentiviral transduction with either an empty vector (EV) or hemagglutinin (HA)-tagged wild-type FANCA. FANCA expression and FANCD2 monoubiquitination were restored by the introduction of FANCA cDNA (Fig. 2D, E). In the MMC sensitivity assay, the complemented cell lines showed a significantly increased survival rate (Fig. 2F). Immunofluorescence showed FANCD2 focus formation in complemented cells (Fig. 2G), and flow cytometry revealed cell cycle recovery in the complemented cell lines after MMC treatment (Fig. 2H).
We performed the MMC sensitivity and FANCD2 monoubiquitination assays to determine the damaging effect of newly detected variants in FA patients. Advances in next-generation sequencing have greatly improved the diagnostic processes in FA. However, a significant portion of FANCA variants remain unclassified (~14%) according to the FA global dataset [8]. This study provides functional evidence for genetic variants associated with FA and can play a decisive role in classifying variants [9].
Go to : 
 XML Download
XML Download