

 PDF
PDF Citation
Citation Print
Print



INTRODUCTION
Immunoglobulin G4-related disease (IgG4-RD) is a systemic, immune-mediated fibroinflammatory condition of unknown etiology characterized by unique histopathological features.1 Once regarded as a disparate single-organ disease, IgG4-RD is now recognized as a multisystem disorder affecting every organ system.1,2 This disease may simultaneously affect several organs. Moreover, it can appear limited to one or two organs and then recur in different organs several years after the onset. Although the reported proportion of organs involved varies widely in the literature, a report found that 60% of cases involved the pancreas, 34% involved the salivary glands, 23% involved the lacrimal glands, 23% involved the kidneys, 20% involved the aorta, 13% involved the biliary tract, 13% involved the lungs, 4% involved the periorbital tissue, and 4% involved the retroperitoneum.3 In addition, the gallbladder, liver, thyroid gland, prostate, and stomach have been reported to be involved. IgG4-RD typically responds well to glucocorticoid treatment. On the other hand, large-scale studies on treatment modalities are needed because of the high long-term recurrence rate.
Autoimmune pancreatitis (AIP) and IgG4-related sclerosing cholangitis (IgG4-SC) are recognized as pancreatic and biliary manifestations of IgG4-RD, respectively, and pancreatic and biliary lesions are the most frequently observed associations. This review discusses the concept, diagnosis, and treatment of AIP and IgG4-SC.
EVOLUTION OF THE CONCEPT OF AIP
1. History of AIP and IgG4-RD
The clinicopathological features of IgG4-RD were first identified in patients with AIP. Sarles et al.4 first described AIP in 1961 when they reported a case of pancreatitis with chronic inflammatory sclerosis and hypergammaglobulinemia. In 1995, Yoshida et al.5 coined the term AIP to describe chronic pancreatitis that improved with glucocorticoid treatment. In 2001, Hamano et al.6 discovered that patients with this particular type of pancreatitis have high serum IgG4 concentrations and suggested that serum IgG4 can provide a valuable means of distinguishing AIP from other pancreatobiliary diseases. In 2003, Kamisawa et al.7 proposed the concept of IgG4-RD based on the similar histopathological features observed in the pancreas, bile duct, gallbladder, liver, and salivary glands of AIP patients. Dr. Kamisawa suggested that AIP is not merely a form of pancreatitis, but rather a pancreatic lesion of an IgG4-related systemic disease that has extensive organ involvement.7
Before the early 2000s, AIP had been described in several articles published in Japan, but few cases have been reported in the United States. As the 2003 clinical paper “Autoimmune Pancreatitis: Does it Exist?” suggested, skepticism about AIP has persisted in the United States,8 and Western doctors implied that AIP may be endemic to Japan. Two years later, it was acknowledged that AIP is a global disease and not confined to Japan.9 As with many diseases, worldwide acceptance and the formal recognition of AIP took even longer.10
2. Development of diagnostic criteria for AIP
In 2002, the Japan Pancreas Society was the first to propose diagnostic criteria for AIP. It revised the criteria in 2006.13 Dr. Chari proposed the HISORt criteria in 2006 and revised them in 2009.14 Dr. Myung-Hwan Kim, a guarantor of this article, proposed the Kim criteria in 2006.15 After the emergence of the need for international consensus standards for the diagnosis of AIP, the international consensus diagnostic criteria (ICDC) for AIP were adopted at the International Association of Pancreatology Meeting in Japan under the leadership of Dr. Shimosegawa and Dr. Chari, and published in 2011.16 In addition to using the ICDC for AIP, Japanese researchers have been periodically revising their diagnostic criteria.17
The ICDC for AIP was established so that there would be unifying criteria available for worldwide application.16 The ICDC for AIP was developed after existing criteria from Japan, the United States, Korea, and Italy were reviewed. The ICDC for AIP was agreed upon by an international panel of experts and is based on five cardinal features of AIP that can be detected by imaging of the pancreatic parenchyma and duct, serology (IgG4), other organ involvement (OOI), histopathology, and response to steroids.
Rheumatology societies do not consider endorsing “diagnostic” criteria; instead, they provide approval for “classification” criteria. The American College of Rheumatology (ACR)/European League Against Rheumatism (EULAR) approved the classification criteria for IgG4-RD in 2019.18 The strengths of the ACR/EULAR IgG4-RD classification criteria are that they include a more detailed classification of the serology and histology, as well as chest involvement of IgG4-RD. On the other hand, the significant issues associated with the ACR/EULAR criteria are their complexity and non-inclusion of focal type AIP. In clinical practice and the research context for AIP, the ICDC for AIP may be more suitable than the ACR/EULAR criteria for IgG4-RD.
3. Pathogenesis of IgG4-RD
With the establishment of diagnostic criteria, diagnosing AIP and IgG4-RD has improved considerably, but the etiology is still not well known. The immune response associated with IgG4-RD may be initiated when an individual sensitive to an unknown immunogen is repeatedly exposed to that immunogen.19 Such unknown immunogens that may trigger IgG4-RD may include self-antigens (e.g., lactoferrin and carbonic anhydrase II), microorganisms (e.g., bacteria), allergens, or occupational antigens (e.g., solvents, industrial gases, or oil products).20,21 When a self-antigen or foreign antigen triggers the immune response, the proportion of follicular helper T (Tfh2) cells increases, and B cells are activated. Tfh2 cells produce transforming growth factor-β, which activates fibroblasts that mediate the extracellular matrix remodeling and tissue damage. They also play a key role in the activation/maturation of B cells and their differentiation into antibody-producing cells.22 Tfh2 cells produce cytokines (i.e., interleukin [IL]-4, IL-10, and IL-21) involved in the immunoglobulin class switch to IgG4.
4. Korean contribution to AIP and IgG4-SC research
The authors’ group has actively contributed to AIP and IgG4-SC research.11,15,23-43 Table 1 lists the significant research contributions to the field of AIP research. The most important contributions include 1) the proposal of Kim's criteria for AIP and 2) the steroid trial to distinguish AIP from pancreatic cancer.44
CLINICAL IMPLICATIONS OF AIP
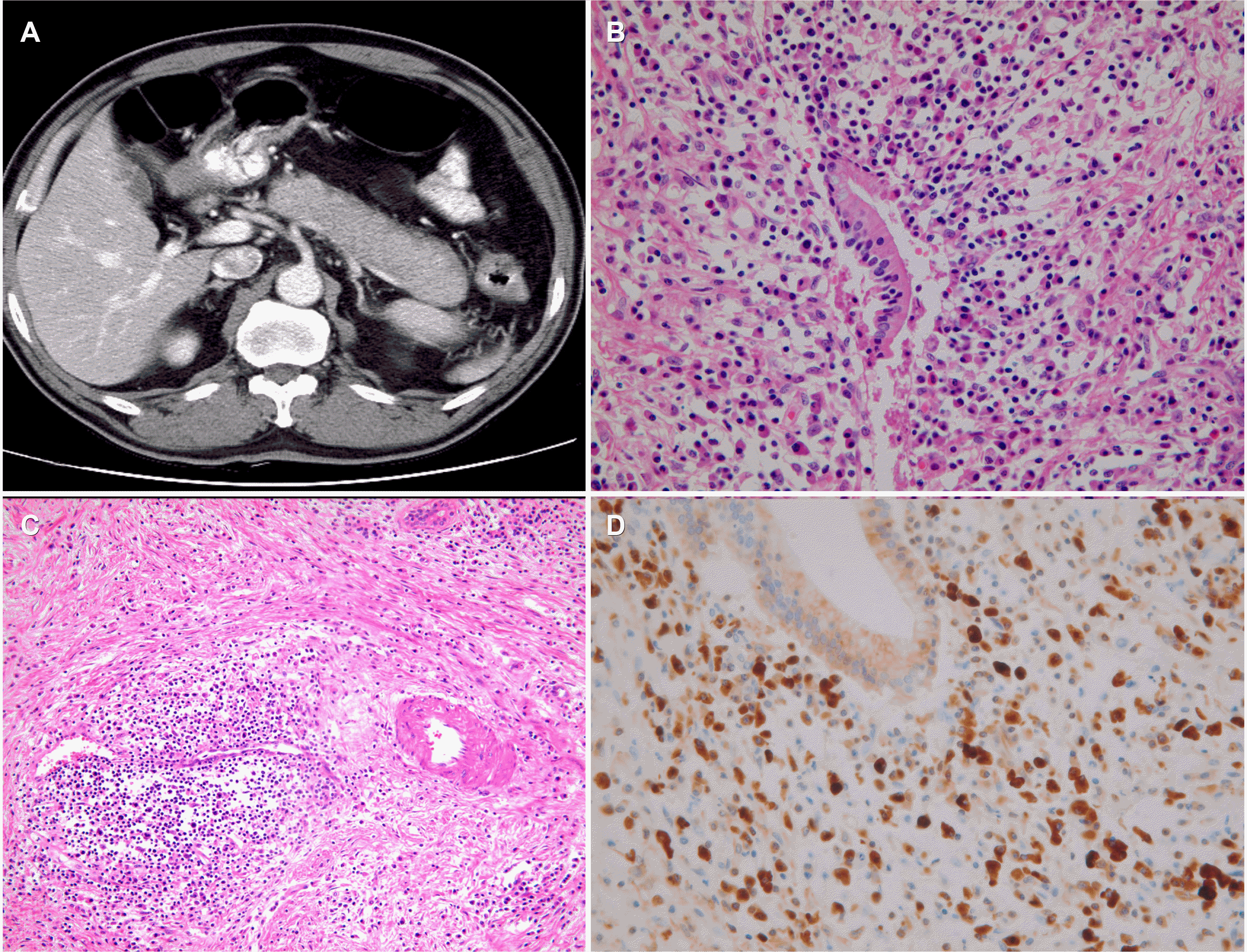
AIP is a unique subtype of pancreatitis; it dramatically responds to glucocorticoid treatment, distinguishing it from ordinary chronic pancreatitis (Fig. 1). When left untreated, IgG4-RD (including AIP) often causes irreversible fibrosis and tissue damage, leading to organ failure. AIP may result in intense fibrosis with calcification/stones, resembling ordinary chronic pancreatitis after multiple relapses. Patients with late-stage AIP may not respond to glucocorticoids. Early diagnosis and appropriate treatment of AIP are essential for a better prognosis.
Differentiating AIP/IgG4-SC from pancreatobiliary malignancies is of utmost importance.45-47 Differentiation between AIP and pancreatic cancer should be based on the combined use of imaging, serology, histopathology, and OOI, because of the lack of a single diagnostic test for AIP. For a diagnosis of IgG4-RD, a finding of no malignant cells by EUS must be paired with radiological information and steroid responsiveness for a diagnosis of IgG4-RD, particularly in patients with indeterminate imaging (e.g., mass-forming focal type).26 Repeat EUS-guided fine needle aspiration (FNA)/fine needle biopsy (FNB) is warranted in patients who demonstrate continued suspicion of pancreatic cancer despite the negative findings in the initial cytology/biopsy.26,32,48 A positive steroid responsiveness might be defined as radiologically demonstrable resolution or marked improvement in main pancreatic ductal narrowing after glucocorticoid therapy and, if present, resolution or measurable reduction of the pancreatic mass.26
There are two main types of misdiagnoses associated with AIP. The first involves AIP being misdiagnosed as pancreatic cancer. In such cases, pancreatic resection may be performed in patients with AIP. The second involves pancreatic cancer being misdiagnosed as AIP. In these cases, glucocorticoids may be administered to patients with resectable pancreatic cancer and then cause a delay that might miss the window for operation.
AIP tends to be both underdiagnosed and overdiagnosed. Underdiagnosis is generally due to a lack of recognition of this disease, and overdiagnosis may result from the overenthusiasm of physicians. Access to a patient’s past medical history is vital for diagnosing IgG4-RD. A thorough history taking and a detailed review of past medical issues often reveal unrecognized manifestations of IgG4-RD. This is because multi-organ involvement can occur metachronously with a wide disease-interval gap. A history of AIP or surgical resection of presumed pancreatic/biliary cancer found to be an inflammatory pseudotumor may provide clues to a diagnosis of IgG4-RD.
DIAGNOSIS OF AIP AND IgG4-SC
1. Imaging features of AIP and IgG4-SC
A recent diagnostic imaging guide for AIP stipulates that, in many cases, AIP cannot be diagnosed accurately using a single modality.49 The most widely used imaging modalities are dynamic contrast-enhanced CT and MRI. CT has a suboptimal sensitivity (59%) and high specificity (99%) for differentiating AIP from pancreatic cancer, whereas MRI has a higher sensitivity (83%) with similar specificity (97%).46
AIP can be suspected by the following imaging features (vs. pancreatic cancer): 1) diffuse pancreatic enlargement with or without a capsule-like rim (vs. parenchymal atrophy above the stricture); 2) delayed homogeneous enhancement of the pancreatic mass (vs. poor enhancement or target-type enhancement); 3) a diffusely attenuated main pancreatic duct with an irregular wall (vs. a single localized stricture); 4) no to mild upstream duct dilatation despite a long stricture (vs. marked upstream duct dilatation); 5) a double duct sign without a pancreatic mass in patients with obstructive jaundice (vs. a discrete pancreatic mass); and 6) OOI unusual for pancreatic cancer, such as the hilar bile duct, salivary glands, kidney involvement, or retroperitoneal fibrosis (vs. liver metastases).26,47
To differentiate AIP from pancreatic cancer, the MRI features favoring AIP are diffuse enlargement (diagnostic OR, 75), a capsule-like rim (52), multiple strictures of the main pancreatic duct (47), and homogeneous delayed enhancement (46).47 In contrast, the MRI features favoring pancreatic cancer are target-type enhancement (diagnostic OR, 41) and a discrete pancreatic mass (35).47
The term “enlargement” of the pancreas can be subjective and vague. The pancreas size can be affected by many factors, including physique and age. For example, an apparently enlarged pancreas can be normal for a large young man. Delayed homogeneous enhancement of the pancreatic parenchyma is another distinguishing feature of AIP. In the arterial phase, the pancreas of a patient with AIP appears hypodense compared to the spleen. In the delayed phase, attenuation increases compared to early images. This reflects fibrosis associated with the inflammatory process.47
Endoscopic retrograde pancreatography (ERP) was once regarded as an essential component for the diagnosis of AIP because it can be used to detect minute changes in the main pancreatic duct.13,50 On the other hand, advancements in MRI, such as the development of 3T units, have resulted in magnetic resonance cholangiopancreatography, generating images of a quality equivalent to ERP images.47
The characteristic imaging findings of IgG4-SC are concentric wall thickening of the bile duct with a smooth luminal surface and preserved luminal patency.45,51 For differentiation between IgG4-SC and cholangiocarcinoma, asymmetrical (eccentric) wall thickening, an irregular luminal surface, and marked proximal duct dilatation may suggest a diagnosis of cholangiocarcinoma.32,48 For the differentiation of IgG4-SC from primary sclerosing cholangitis, the cholangiographic findings of the beaded appearance is mostly indicative of PSC, but older age and OOI mostly indicate IgG4-SC.42
2. Biopsy diagnosis of AIP
The key histopathological features of type 1 AIP include 1) diffuse lymphoplasmacytic infiltration and fibrosis, 2) numerous IgG4-positive cells, 3) storiform fibrosis, 4) obliterative phlebitis, and 5) ductal lesion (periductal infiltrates with fibrosis). 16,41,52 EUS-guided sampling from the pancreas can provide a definite diagnosis of AIP. Endosonographers use FNB needles for patients with suspected AIP because acquiring sufficient amounts of tissue is essential for an AIP diagnosis.41,52 A recent meta-analysis showed that the pooled diagnostic yields for level 1 or 2 histology criteria of AIP were 55.8% for FNA and 87.2% for FNB (p=0.030).41 Therefore, FNB needles (≥22 G in size) should be used when a pancreatic biopsy is required to differentiate AIP from pancreatic cancer.
Regarding the pathological examination of samples, agreement among pathologists about the diagnosis of type 1 AIP has been reported to be suboptimal.52 The observer-dependent nature in interpreting storiform fibrosis and obliterative phlebitis is problematic.41,52 For a better evaluation of obliterative phlebitis, the addition of elastic stains, such as Elastica van Gieson staining, should be considered.41 IgG4 immunostaining of pancreatic tissue has a reported sensitivity and specificity of 70% and 92%, respectively.39 In addition, a biopsy sample from the bile duct or ampulla stained for IgG4 can assist in diagnosing AIP owing to its moderate sensitivity and high specificity.29,39 Hence, a multi-disciplinary collaborative approach that involves specialists in gastroenterology, radiology, and pathology is needed to reach a diagnosis of AIP.52
TREATMENT AND RELAPSE OF AIP
Glucocorticoids are the first-line therapy for AIP and IgG4-RD. The indications for glucocorticoid treatment are symptoms, such as obstructive jaundice, abdominal pain, back pain, and the presence of symptomatic extrapancreatic lesions.17 The treatment goal may be to achieve symptomatic and radiological remission. Normalization of the serum IgG4 may not be achieved after clinical/radiological remission.40 The recommended initial oral prednisolone dose for the induction of remission is 0.6 mg/kg/day, which is administered for 2-4 weeks, followed by a gradual taper to a maintenance dose.17 On the other hand, despite experiencing dramatic remission after glucocorticoid therapy, patients with type 1 AIP continue to have a high likelihood of relapses over a long period.40 Relapses may be symptomatic, radiological, serological, or histological, and analogous to remission.53
The suggested risk factors of relapse are high serum IgG4 levels before treatment, persistently high serum IgG4 levels after steroid treatment, diffuse enlargement of the pancreas, proximal IgG4-SC, and extensive multi-organ involvement.35 To prevent relapse, long-term maintenance therapy (approximately three years) with low-dose prednisolone can be applied.40 A dose of 5 mg/day or more may be preferred over a dose of 2.5 mg/day to prevent relapse.54 For relapsed AIP, the re-administration of a high dose of glucocorticoids with a slow steroid taper is effective. In patients with multiple relapses or severe steroid intolerance, steroid-sparing agents can be considered, including immunomodulators or rituximab.
1. Monitoring disease activity
In general terms, an ideal serological biomarker provides information that is helpful during diagnosis, while monitoring the disease activity, when predicting the prognosis, and when tracking the response to therapy. The serum IgG4 has limited use in monitoring the disease activity of AIP, even though it is a useful biomarker for diagnosing type 1 AIP. A one-fold cut-off value of the serum IgG4 (140 mg/dL) has a sensitivity and specificity of 72% and 93%, respectively. In contrast, a two-fold cut-off value has a sensitivity and specificity of 43% and 98%, respectively.55 Peripheral blood plasmablasts count may be a promising biomarker of IgG4-RD. According to Wallace et al., circulating plasmablast counts may be a surrogate marker with greater sensitivity and specificity for diagnosing IgG4-RD.56 The plasmablast counts may also be a potentially useful biomarker for assessing the response to therapy and determining the time to re-treat patients.56
The IgG4-RD responder index (IgG4-RD RI) was published in 2012 and is used to quantify the disease activity and the therapeutic effects of treatments for IgG4-RD.57 An international validation study on the IgG4-RD RI in 2018, which involved 26 international experts, found that the IgG4-RD RI is a good tool for evaluating the disease activity owing to its high reliability.58 Nevertheless, considerable effort is required to evaluate the activity of various organs throughout the body, which may limit its clinical utility.
RESEARCH PROSPECTS
Although the diagnosis and treatment of AIP have benefited from the expanding knowledge of AIP and IgG4-RD, there is a need for further research. Identification of a better biomarker beyond serum IgG4, which can be used as an indicator for diagnosis, disease activity, and response to treatment, is required. The initiating antigens and events that trigger immune-mediated inflammation must be identified, as well as the pathogenic mechanisms and pathways. The roles of B cells, T cells, and other immune cells in the initiation and progression of inflammation and fibrosis should also be investigated. Finally, developing more effective drugs for remission maintenance and relapse prevention is warranted.
 XML Download
XML Download