

 PDF
PDF Citation
Citation Print
Print



INTRODUCTION
C1q nephropathy is an uncommon type of glomerulonephritis that was first described by Jennette and Hipp [1] in 1985 [2-5]. It is characterized by extensive and dominant C1q mesangial deposition in the absence of systemic lupus erythematosus [2,4,5]. Its clinical and microscopic presentations are varied and diagnosis is based on histopathology [2,6]. Typically, patients show proteinuria, and the condition is relatively resistant to steroid treatment [2,6]. Despite decades of studies, a limited number of cases have been reported and C1q nephropathy is still poorly understood. The prognosis is generally favorable except in patients with nephrotic syndrome and focal segmental glomerulosclerosis [2].
There are even fewer studies about C1q deposition in transplanted renal allografts [5,6]. Said et al. [6] reported a series of cases with C1q deposition in renal allografts. Only half of the patients had proteinuria, of whom 17% had proteinuria >1 g/day. The 1-year follow-up showed that most patients had stable creatinine levels, with no or stable proteinuria [6]. However, the clinicopathologic presentations and outcomes in transplanted kidney allografts have yet to be elucidated [5]. The purpose of this study was to retrospectively review and describe the clinical presentations, histopathology, and outcomes of kidney-transplanted patients with C1q deposition in the renal allografts.
METHODS
The study was approved by the Institutional Review Board of Seoul National University Hospital (SNUH; IRB No. 2112-105-1284). The need for informed consent was waived due to the retrospective nature of the study.
Patient Selection and Data Collection
A retrospective review of kidney transplantations performed at SNUH between January 2005 and December 2018 was undertaken. Patients with both protocol and indication renal allograft biopsies were included, and patients whose specimens displayed C1q deposition were further selected. These patients’ electronic medical records were comprehensively reviewed to extract data on clinical variables, the type of transplantation, time since transplantation, the cause of end-stage renal disease, laboratory findings, immunosuppressive therapies, and infection history. Long-term patient outcomes, including serum creatinine, urine protein and creatinine, allograft survival, and patient survival at last follow-up were also recorded. Nephrotic-range proteinuria was defined as a spot urine protein to creatinine ratio >3.5 mg/mg, while a ratio of less than 0.2 mg/mg was defined as normal.
Immunosuppression
All patients were prescribed a triple-immunosuppressant regimen composed of tacrolimus, mycophenolate mofetil, and a steroid (prednisone). During the first 3 months postoperation, the tacrolimus trough level was targeted to 8–12 ng/mL, followed by a concentration of 6–10 ng/mL until 1 year after transplantation and 4–6 ng/mL thereafter. Mycophenolate mofetil was administered at a fixed dose of 500 mg twice a day. An intravenous dose of 10 mg/kg of prednisone was administered during the operation, followed by rapid tapering to achieve a daily dose of 5 mg within the first month after transplantation. As induction immunosuppression, basiliximab (20 mg) was administered on days 0 and 4 after transplantation. In high-risk patients with high panel-reactive antibodies (>80%), preformed donor-specific antibodies (DSAs), and/or ABO-incompatibility, induction with anti-thymoglobulin (1.5 mg/kg/day for 0–3 days) was administered instead of basiliximab.
Pathologic Findings
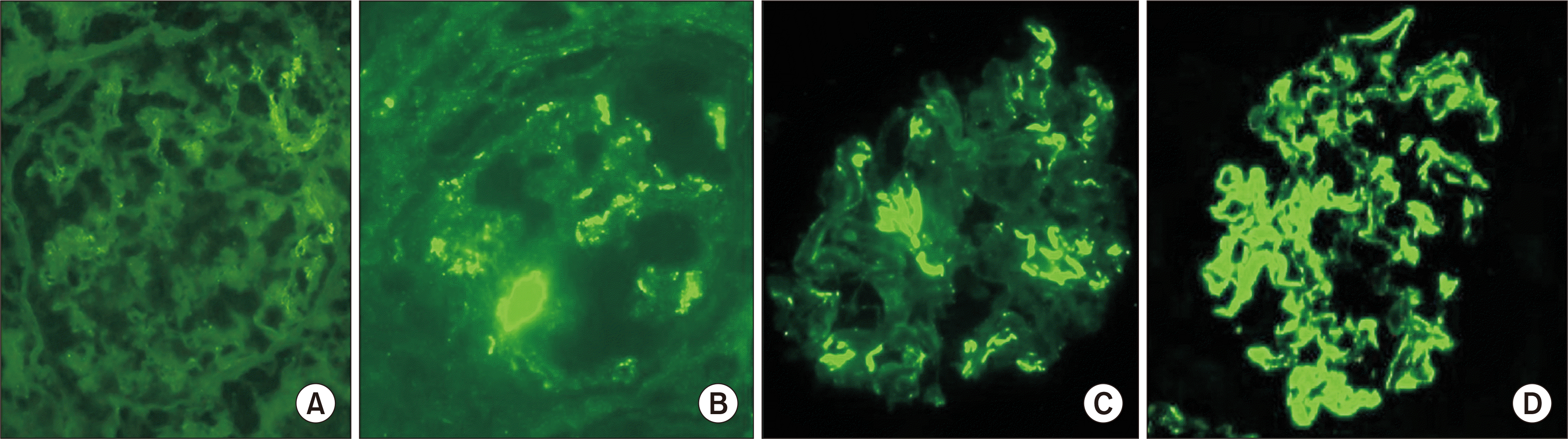
Protocol biopsies at SNUH include a reperfusion biopsy, defined as a biopsy performed immediately after reperfusion, a postoperative 2-week biopsy, and a postoperative 1-year biopsy. All other biopsies are performed when indicated due to worsening of the graft, defined as a greater than 20% increase in baseline serum creatinine, and/or an appearance of de novo anti-human leukocyte antigen DSAs. All biopsy specimens were processed using standard protocols for light microscopy, immunofluorescent microscopy (IF), and electron microscopy. The degree of IF staining was graded as – (negative), ± (trace), 1+ (mild), 2+ (moderate), 3+ (marked), and 4+ (heavy) according to Jennette’s criteria [1]. IF stains with less than 2+ staining were excluded in this study. Immuno-complex depositions without a well-phenotype autoimmune disorder were classified as Immune-complex glomerulopathy not otherwise specified, as suggested by Chin et al. [5]: full-house (+IgG/+IgM/+IgA/+C3/+C1q), quasi-full house (+IgG/+C3/+C1q), IgA-rich (IgA dominant/codominant), and C1q-rich (–IgG/+C1q). The specimens were classified in accordance with the Banff 2007 and 2013 classifications by experienced renal pathologists (KCM). C1q disappearance was defined as negative IF staining on a follow-up biopsy. Diminished C1q was defined as diminished IF staining for C1q to either trace or 1+ (mild) in a follow-up biopsy.
Statistical Analysis
The statistical analysis was conducted using IBM SPSS ver. 21.0 (IBM Corp., Armonk, NY, USA). All tests were two-tailed and differences at P-values <0.05 were considered statistically significant. Differences in mean values between groups were compared using the Student t-test, and categorical variables were compared using the chi-square test or Fisher's exact test.
RESULTS
Eligible Cases
A total of 1,742 kidney transplantations were performed at SNUH from January 2005 to December 2018. All protocol biopsies, including the renal reperfusion biopsy, were performed in 1,357 of these cases. All renal allograft biopsies of these patients were retrospectively screened for C1q deposition (n=10,217). C1q deposition was detected in 104 cases (3.6%, n=367/10,217 biopsies), of which only 28 (1.6%, n=58/367 biopsies) had intense (≥2+) C1q deposition and were finally reviewed in this study (Fig. 1).
Histopathologic Findings
The mean number of glomeruli sampled was 8.0 (standard deviation, 3.6). Proliferative glomerular changes were found in four (14.3%) cases and the remaining 24 (85.7%) of the cases did not have any other lesions indicative of glomerular injury. Other findings on light microscopy accompanying C1q deposition were interstitial fibrosis and tubular atrophy in 11 cases (39.3%), followed by 9 (32.1%) cases of borderline or acute cellular rejection and 1 (3.6%) case of mixed acute cellular and antibody-mediated rejection. There were no cases of antibody-mediated rejection. Three patients (10.7%) had histological evidence of BK polyoma nephropathy and five (17.8%) showed immunoglobulin A (IgA) nephropathy; these cases were de novo IgA nephropathy, except in one patient (Table 1).
On electron microscopy, electron-dense deposits were present in 92.3% (n=26), and four of the cases showed marked podocyte foot effacement. In the analysis of immune complex subtypes, the most frequent type seen was the C1q-rich type (n=20, 71.4%) without IgG deposition, followed by full-house (n=4, 14.3%), quasi-full house (n=2, 7.1%) and the IgA-rich subtype (n=2, 7.1%).
On immunofluorescence microscopy, staining for C1q was 2+ in 85.7% (n=24/28) of the cases and 3+ in the remaining four cases. There were no cases of 4+ staining for C1q deposition. The most frequently accompanying intense (≥2+) stains were IgA (n=11), followed by C3 (n=10), lambda (n=6), kappa and IgM (n=4 each) and lastly, IgG (n=3) (Table 2).
Clinical Data and Characteristics
The mean age was 37.6±17.1 years and 71.4% of the patients were male. None of the patients had underlying C1q nephropathy or other types of auto-immune glomerulonephritis, such as systemic lupus erythematosus. Among the 28 cases, 18 cases were from living donors, half of whom were male with a mean age of 46.4±11.5 years.
C1q deposition was initially detected during protocol biopsy in 12 cases (42.9%) and during indication biopsy in 16 cases (57.1%). All except two patients in the protocol biopsy group had initial detection of C1q deposition during the reperfusion period. In these two patients, C1q deposition was detected in postoperative 2-week and 1-year protocol biopsies. None of the patients in the indication biopsy group had C1q detection in any previous biopsies, including the protocol biopsies. The mean time to initial C1q deposition in the indication biopsy was 1,529±1,254.8 days. The baseline characteristics were similar in both groups, with a mean follow-up of 7 years.
The transplant characteristics were similar, except that preoperative desensitization was more frequent in the protocol biopsy group (P=0.034). This was due to pre-formed DSAs (n=2) and ABO incompatibility (n=1). The protocol biopsy group had worse initial serum creatinine levels (P=0.008) and estimated glomerular filtration rate (eGFR) at initial detection of C1q (P=0.002). However, after 3 years of follow-up, serum creatinine and eGFR showed no significant difference (Table 3).
Follow-up Biopsy Findings
Follow-up biopsies were only performed in 15 cases (53.6%) (Table 4), including all of the protocol biopsy cases (n=12), but only three of the indication biopsy cases. The C1q depositions either disappeared (n=13, 86.7%) or diminished (n=2, 13.3%). The median interval to C1q diminishing or disappearance was 402 days (range, 8–2,128 days). Two patients (13.3%) had borderline or acute cellular rejection on the follow-up biopsy and three patients (20%) showed interstitial fibrosis and tubular atrophy. There were two cases (13.3%) of BK polyoma nephropathy.
DISCUSSION
The prevalence of dominant or codominant C1q deposition in transplanted renal allograft biopsies was 1.6% (n=28 of 1,742 total cases). None of the 28 patients reviewed had underlying C1q nephropathy in the native kidney. However, the 10 cases of C1q deposition detected on reperfusion biopsies should be interpreted with caution. Since reperfusion biopsy is undertaken immediately after reperfusion of the renal allograft, the C1q deposition in this group could signify the presence of already existing C1q deposition in the donor allograft. The retrospective design of this study makes it impossible to know the time of appearance of C1q deposition in renal allografts. The remaining 18 cases (1.0%) could more accurately reflect the prevalence of C1q deposition in transplanted renal allografts. While the reported prevalence of C1q nephropathy in the general population ranges from 0.2% to 16%, the reported prevalence in transplanted kidneys is lower, ranging from 0.01% to 3% [1,2,4-7]. Despite this, along with focal segmental glomerulosclerosis and membranous glomerulonephritis, C1q deposition still accounts for one of the three most common de novo morphological glomerular patterns in renal allografts [5,6].
Except in one case that showed immune complex-mediated proliferative glomerulonephritis, there were no other accompanying glomerular changes. Upon review of other immune complexes on IF staining, the most common type of immune complex was that of the C1q-rich subtype. In other studies, C1q was most frequently found with IgM and IgG, as they provide ligands for C1q immune complex formation [2,5,8]. The absence of other immune complexes in IF staining may support Kanai’s hypothesis that C1q might only be an innocent bystander, like IgA deposition, which is found in 4.8% of the general population, while the incidence of IgA nephropathy is only 0.02% [4]. The spontaneous disappearance or diminishing of C1q deposition may further support this hypothesis. Stable serum creatinine levels with stable or improved proteinuria were also seen. This is similar to other reports, and as Said et al. [6] have suggested, the mild clinical and pathological features may be due to early disease detection and the therapeutic effects of maintenance immunosuppression regimens or rejection treatment.
The most common hypothesis of C1q nephropathy is the post-infection activation of the inflammatory cascade with subsequent complement activation, consumption, and accumulation resulting in binding of C1q to other immunoglobulins and becoming entrapped in the paramesangial region and increasing mesangial trafficking [1,2,7,9]. In this study, three cases of BK nephropathy with C1q deposition showed disappearance of both BK nephropathy and C1q deposition on follow-up biopsies. Similarly, half of the patients in the report by Said et al. [6] had preceding infections, but this prevalence of infection was not higher than that in the general renal transplant recipient population. The authors [6] further argued that the absence of endocapillary hypercellularity or weaker staining for C3 than for C1q or the absence of subepithelial humps constitutes evidence against the possibility of such a hypothesis. Other authors [4,8,10] have suggested that C1q nephropathy may overlap with or be superimposed upon other glomerulopathies. However, the results of this cohort showed only one case of immune complex–mediated proliferative glomerulonephritis, and upon review of other immune complexes on IF staining, the most common type of immune complex was that of the C1q-rich subtype.
C1q deposition in the indication biopsy group was frequently accompanied by T-cell mediated rejection or interstitial fibrosis and tubular atrophy. This may have been the result of selection bias, as the indication biopsies were performed undertaken due to either a worsening of graft function or an appearance of de novo DSAs. Some studies have proposed that C1q-mediated antibody rejection leads to poorer allograft outcomes; however, the presensitized patients were all in the protocol biopsy group and not in the indication biopsy group, and none had antibody-mediated rejection [6,10].
Although limited in number, as shown by the stabilization in serum creatinine levels and proteinuria and the disappearance of C1q deposition, one could conclude that the presence of C1q deposition itself does not influence the graft failure rate and is most likely clinically silent. Similarly, other reports on C1q deposition or other immune complex depositions concluded that glomerular changes due to immune complex deposition are clinically silent and do not influence graft function [4-6,8]. However, proteinuria after kidney transplantation is associated with reduced graft survival, independent of glomerular pathology, graft function, and rejection [6]. Therefore, if a patient presents C1q deposition and proteinuria, active measures are recommended.
To our knowledge, this study reports the largest number of C1q deposition cases in transplanted renal allografts to date. Although a rare and poorly understood disease, the results from this cohort and other (few but existing) cohorts suggest that C1q deposition is most likely clinically silent and may be an incidental finding. The prevalence of dominant or codominant C1q deposition in transplanted renal allograft biopsies was 1.6%. Most cases did not have any other accompanying glomerular changes. The follow-up biopsies of these allografts showed spontaneous disappearance or diminished staining of C1q deposition. Due to the limited number of cases, firm conclusions about renal allograft outcomes could not be made. However, the findings of this study suggest that C1q deposition found in renal allografts is most likely clinically benign.
 XML Download
XML Download