

 PDF
PDF Citation
Citation Print
Print



INTRODUCTION
Primary liver sarcomas are very rare, and they represent less than 0.1% of all primary liver tumors [1,2]. They are a heterogeneous group of malignancies with their biology ranging from indolent behavior of epithelioid hemangioendothelioma with a 5-year survival of 55%–75% after transplantation or resection to aggressive angiosarcoma associated with dismal prognosis [2-4]. Rhabdomyosarcoma is the most common type of soft-tissue sarcoma in children [5]. Only 1% of pediatric rhabdomyosarcomas affect the biliary tract. Biliary rhabdomyosarcoma is a rare tumor, but it is still the most common tumor of the biliary tract in children [6]. Treatment with chemotherapy and surgery achieves 5-year survival rates greater than 75% in patients with nonmetastatic disease [7]. Complete resection of biliary rhabdomyosarcoma is rarely possible despite aggressive surgery. Gross residual disease after surgery is associated with poorer disease-free survival than complete excision of the tumor [7-9].
The optimal treatment approach for hepatic sarcomas is not yet well defined. Surgery is the only treatment modality that can offer a potential cure. However, the existing studies of outcomes after resection of hepatic sarcomas are limited by the small number of patients treated over extended periods of time [6,7]. Among the patients who undergo surgery, the indications for surgical resection versus transplantation are not defined. Moreover, the benefit of adjuvant treatment remains questionable with the existing literature reporting minimal benefit [2]. Liver transplantation is an effective treatment for children with unresectable liver tumors [6,10,11]. To the best of our knowledge, only two cases of liver transplantation have been reported in children with biliary rhabdomyosarcoma [12,13].
We herein report the case of a 6-year-old boy with biliary embryonal rhabdomyosarcoma and liver metastasis, which was treated with neoadjuvant and adjuvant chemotherapy combined with living donor liver transplantation (LDLT).
CASE REPORT
This study was approved by the Institutional Review Board of Asan Medical Center (IRB No. 2020-0836), which waived the requirement for informed consent due to the retrospective nature of this study.
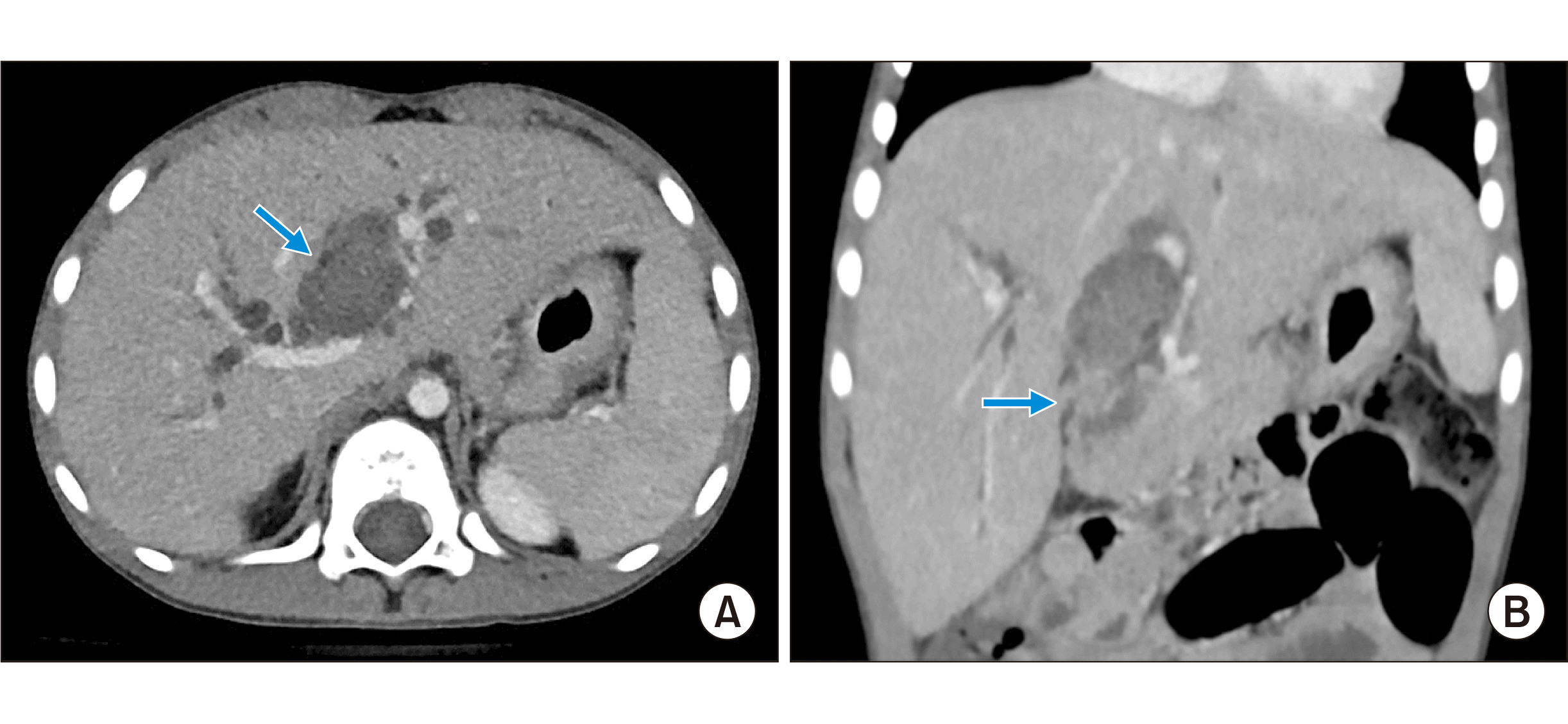
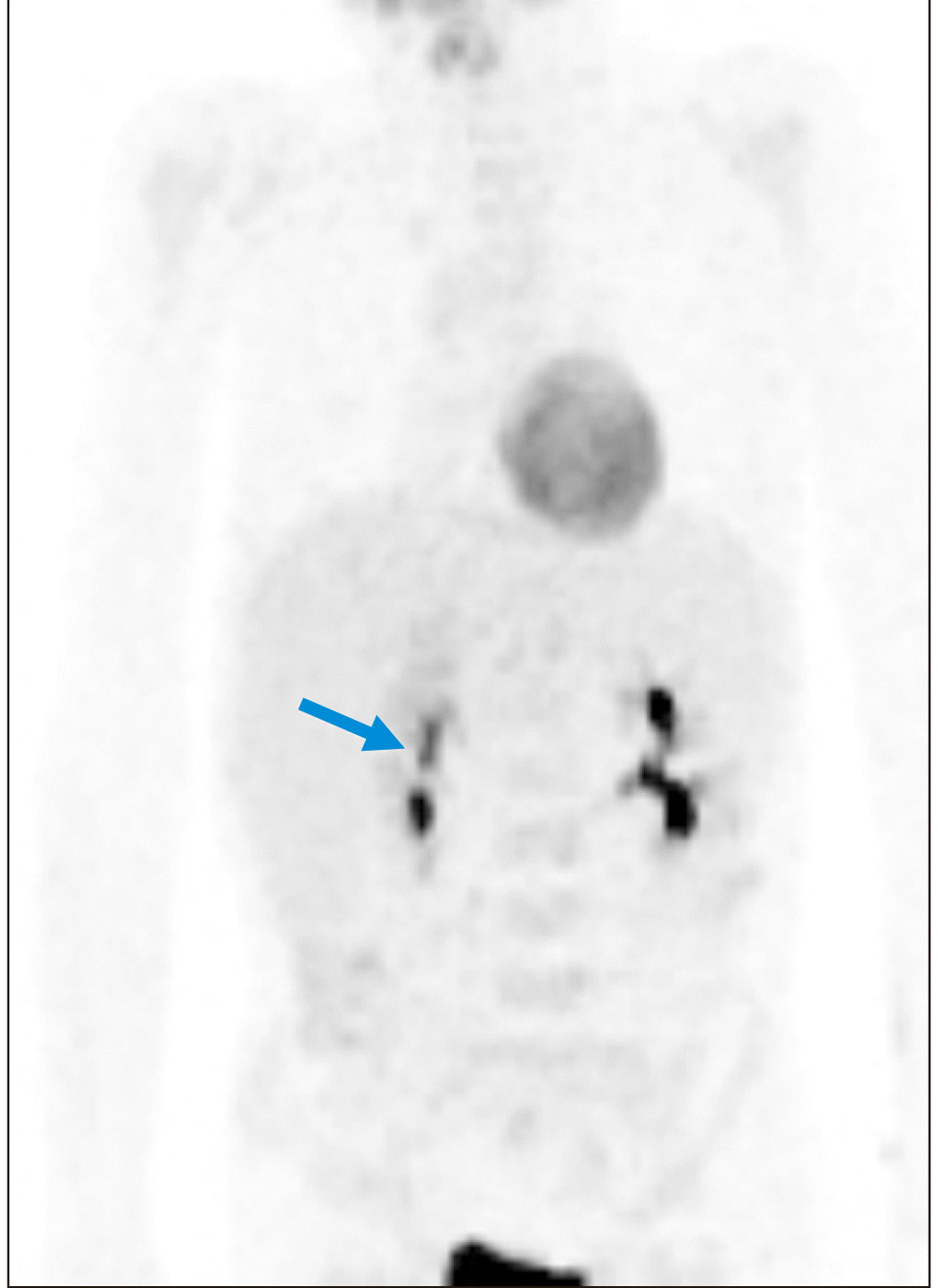
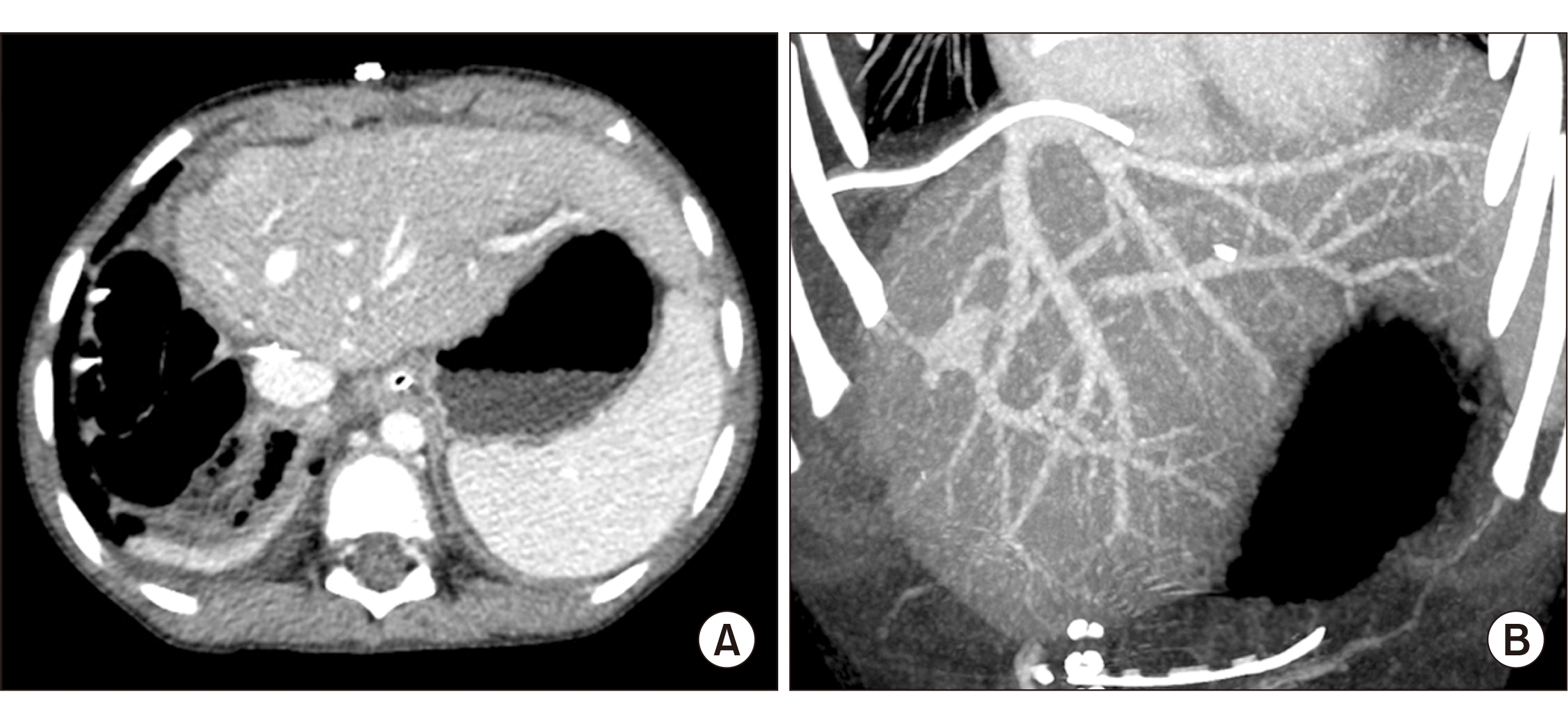
A previously healthy 6-year-old boy who presented with jaundice was referred to our institution for work-up. Serum total and direct bilirubin levels were 3.7 mg/dL and 3.2 mg/dL, respectively. Computed tomography (CT) scan showed a low-attenuation intraductal mass from the left hepatic duct to the intrapancreatic common bile duct with diffuse upstream dilatation of the intrahepatic duct and multiple liver metastases (Fig. 1), which suggested tumorous conditions such as rhabdomyosarcoma. Endoscopic ultrasonography-guided fine needle aspiration biopsy of the bile duct confirmed the pathological diagnosis of rhabdomyosarcoma. Whole body fluorodeoxyglucose-positron emission tomography revealed a hypermetabolic mass in the left hepatic duct and common bile duct, suggesting a biliary tract tumor (Fig. 2). Bone marrow biopsy revealed no metastasis of rhabdomyosarcoma.
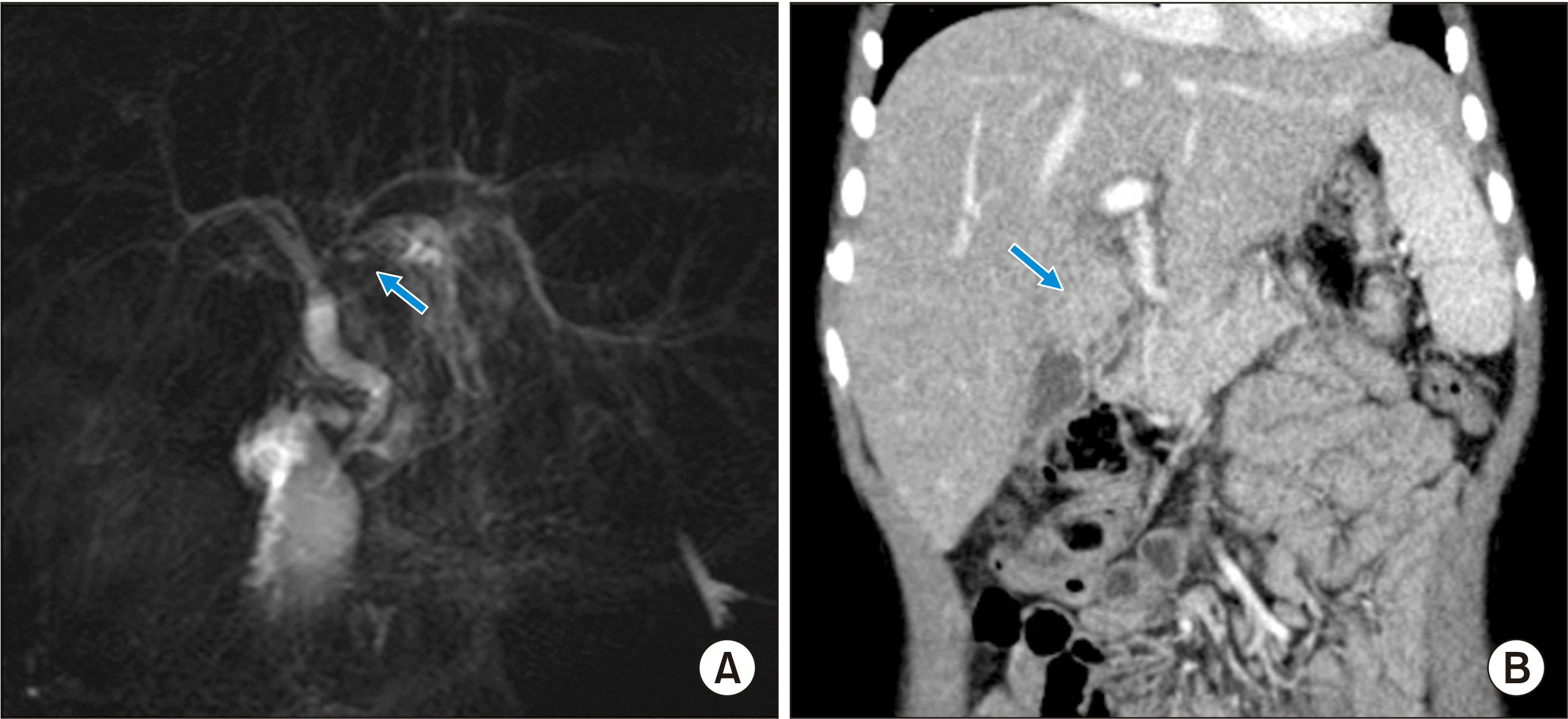
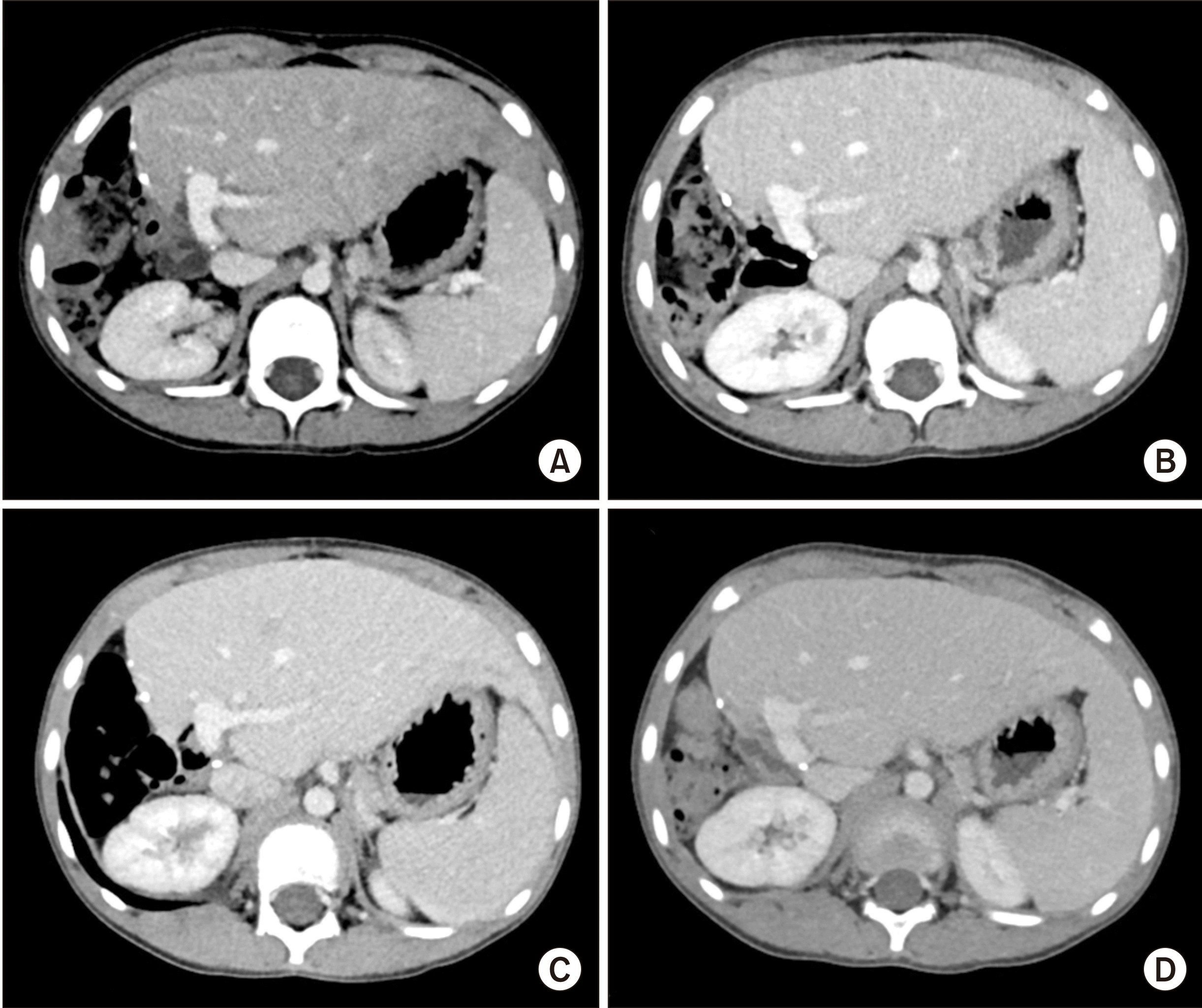
After insertion of a chemoport, induction chemotherapy for intermediate risk rhabdomyosarcoma was started [14]. Intraductal rhabdomyosarcoma and intrahepatic metastases were reduced after systemic chemotherapy for 3 months (Fig. 3), and obstructive jaundice was resolved. Although significant reduction of tumor extent was achieved, it was still beyond the extent of surgical resection. Thus, LDLT was planned to remove the tumor completely. LDLT operations in both the donor and recipient were performed according to the routine procedures. A left lateral section graft weighing 330 g was harvested from his 38-year-old mother, and the graft-to-recipient weight ratio was 1.94%. The left hepatic vein orifice on the graft was enlarged with patch venoplasty according to our institutional guidelines.
During recipient hepatectomy, the distal common bile duct was excavated within the intrapancreatic portion to remove the tumor-involved distal bile duct completely. The distal bile duct resection margin was tumor-negative on frozen-section biopsy. The hepatic hilum was meticulously dissected to prevent potential tumor spread. The remaining part of the recipient hepatectomy was performed according to the standard procedures of pediatric LDLT. The left lateral section graft was implanted according to the routine procedures of pediatric LDLT, including size-matched anastomoses of the graft hepatic vein and portal vein, single left hepatic artery reconstruction using surgical microscopy and Roux-en-Y hepaticojejunostomy.
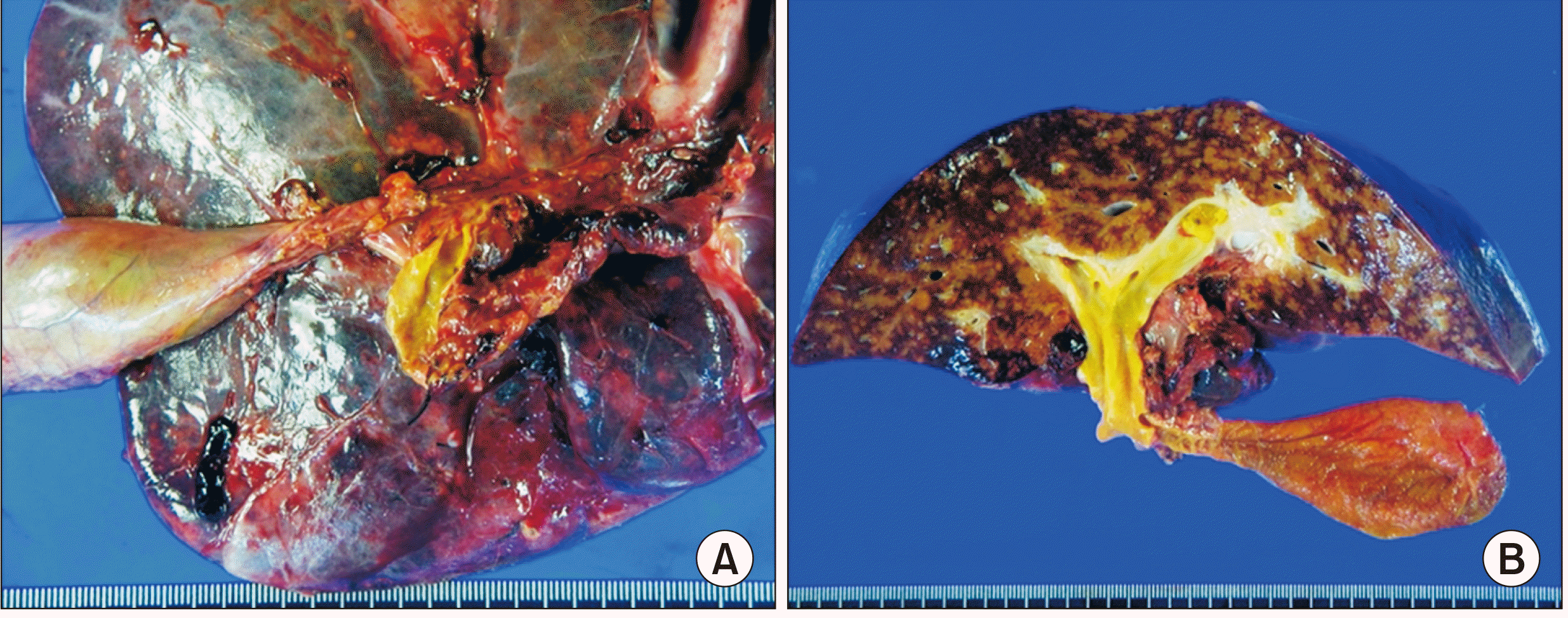
The explant liver showed a minimal residual embryonal rhabdomyosarcoma with no metastasis in the three regional lymph nodes (Fig. 4). The liver parenchyma showed mild fatty change, multifocal sinusoidal dilatation and congestion. Periportal, perivenular and perisinusoidal dilatation were noted. These findings were suggestive of features of the chemotherapeutic effect. The patient recovered uneventfully from the LDLT operation. Early posttransplant CT showed no abnormal findings (Fig. 5). At 1 month after LDLT, scheduled chemotherapy using vincristine, actinomycin and cyclophosphamide was performed for 6 months. At 22 months after LDLT, the chemoport was removed. Follow-up CT scans showed no evidence of tumor recurrence for 26 months after LDLT (Fig. 6).
DISCUSSION
Biliary rhabdomyosarcoma typically occurs in children and infants, and the average age of diagnosis is 3.5 years. The common manifestations usually include jaundice, abdominal pain, bloating, vomiting, and fever [15]. Compared with rhabdomyosarcoma at other sites, rhabdomyosarcoma of biliary pathology usually suggests embryonic origin and therefore is more sensitive to chemotherapy [15]. However, because rhabdomyosarcoma is extremely aggressive, complete excision cannot be achieved in most patients. Moreover, postoperative pathology showed only approximately 8% of negative tumor margins [9]. However, previous studies have reported that among children with positive tumor margins, 92% of these patients treated with postoperative chemotherapy had no tumor recurrence [9]. However, the therapeutic value of surgery in biliary rhabdomyosarcoma is controversial.
Tumor recurrence is an important risk factor of poor prognosis in patients with rhabdomyosarcoma, based on the fact that the 5-year survival rate is only 0% to 17% in these patients [9]. In a case report with salvage liver transplantation after surgical resection [13], the recurrent tumor occupied the hepatic hilum and could not be completely removed. Radiographic assessment showed no lymph node infiltration or distant metastasis; therefore, liver transplantation was performed to completely remove the whole tumor. Postoperative follow-up studies showed no tumor recurrence, with significantly improved quality of life in this patient. Another case report also presented a 4-year disease-free survival after liver transplantation and chemotherapy, suggesting that when coupled with effective chemotherapy, liver transplantation might be considered a potential treatment for unresectable biliary rhabdomyosarcoma [14].
It is known that long-term use of immunosuppressive agents after liver transplantation may lead to the possibility of tumor recurrence or neoplasm. Children with a history of biliary rhabdomyosarcoma who received liver transplantation were given tacrolimus, mycophenolate mofetil, and steroids, without any tumor recurrence at 4 years after surgery [14]. In another case, the patient was given tacrolimus and steroids, with early withdrawal [13]. The concentration of tacrolimus was maintained at a lower level than that in the usual pediatric recipients and reduced doses of chemotherapy drugs were also given. For pediatric patients who underwent liver transplantation for biliary rhabdomyosarcoma, reducing the amount of immunosuppressant and chemotherapy drugs, with close follow-up, ensures maintenance of normal graft function and decreases the side effects of immune suppression and chemotherapy.
In conclusion, liver transplantation could be an effective treatment for unresectable biliary rhabdomyosarcoma in children, according to the location of the tumor. Liver transplantation completely removes the primary tumor, reduces the long-term side effects of chemotherapy, and improves the survival rates of patients, with a good quality of life. The risk of liver transplantation has to be balanced against the chemo-responsiveness of the tumor. Rigorous preoperative evaluation and close postoperative follow-up are important to ensure that pediatric patients with biliary rhabdomyosarcoma benefit from liver transplantation.
 XML Download
XML Download