

 PDF
PDF Citation
Citation Print
Print



INTRODUCTION
Gliomas comprise about one third of all brain tumors and more than three quarter of all malignant brain tumors [1,2,32]. It is considered as one of the most aggressive human malignancies [5,14]. The prognosis of glioma patients is extremely poor due to lack of approaches for early-diagnosis [18,24]. And rapid progression of glioma also greatly contributes to its dismal outcomes [26,38]. Currently, molecule-targeted therapeutic strategy has become the focus of glioma research, and showed promising prospects, which leads to an urgent demand for novel therapeutic targets of glioma.
EID3 (EP300-interacting inhibitor of differentiation) was identified as a novel member of EID family, which inhibits CREB binding protein (CBP)-dependent co-activation [3]. Previous studies revealed that it could inhibit cellular differentiation by binding to class I histone deacetylases (HDAC) or CBP/P300 complex [3,15,28]. A pilot study reported that EID3 serves as a key regulator of cancer stem cell in colorectal cancer [27]. With decreased proteasome activity, EID3 is transcriptionally up-regulated, resulting in activation of Wnt-β-catenin signaling pathway, which plays a vital role in proliferation, invasion, and self-renew of colorectal cancer stem cells [27]. However, the function of EID3 in glioma cell remained elusive.
AMP-activated protein kinase (AMPK) is identified as a master regulator of energy and metabolism with a highly-conserved trait [4,23]. Moreover, AMPK can restore energy balance during metabolic stress [31,39]. Previous investigations reported that AMPK acted as a vital suppressor in cancer through various ways [7,36,41]. Activated AMPK could inhibit the activation of mechanistic target of rapamycin complex 1 (mTORC1), which is considered as a key regulator of oncogenic cascade in tumor development and progression [8,10,16]. Moreover, recent studies revealed that activated AMPK could trigger autophagy to inhibit cancer cell proliferation and induce cell death [19,25]. Additionally, AKT-MDM2-Foxo3 cascade is blocked after AMPK activation [9]. AMPK was also reported to be highly suppressed in glioma, and restoration of AMPK expression in glioma showed promising therapeutic potential [40]. The identifying of AMPK as a novel key upstream regulator in glioma might imply a novel strategy to glioma therapy. We also search AMPKα1, one of major regulators in AMPK signal pathway, in the Cancer Genome Atlas data set and found that it was a mutant gene in the pathogenesis of glioma, and the mutation rate was 57.14%. The type of mutation is substitution, and the result of mutation is missense.
In this study, we explored the function of EID3 in glioma cell proliferation, survival, death, and invasion. Our data identified EID3 as a novel upstream inhibitor of APMK, which provide a new insight into the mechanism of glioma progression and indicated that EID3 might be a promising target for glioma therapy.
MATERIALS AND METHODS
The protocols of using human cells and tissues were in accordance with the principle of Declaration of Helsinki, and were approved by the Ethics Review Board (ERB) of Minhang Hospital.
Cell culture
A172 glioma cell line was purchased from the Cell Bank of Chinese Academy of Sciences (Shanghai, China). A172 cells were cultured in DMEM medium (Gibco, New York, NY, USA) supplemented with 10% fatal bovine serun (FBS) (Invitrogen, Carlsbad, CA, USA), 100 U/mL penicillin and 100 µg/mL streptomycin (Gibco) at 37°C in a humidified incubator (Thermo-Fisher, Waltham, MA, USA) with 5% CO2. All experiments were conducted with cells passage less than eight times. DNA fingerprinting and profiling were performed every 5 months to verify the cell line phenotype. Human astrocyte culture was performed according to previous studies [11,21].
Clinical glioma tissues
A total of five patients with glioma were recruited in present study (male, World Health Organization grade III, age 54–68) at Minhang Hospital. Right after surgery, tumor tissues and surrounding normal brain tissues were collected, and separated carefully under microscope. Glioma was defined by pathological tests with both fresh frozen section tissue and formalin-fixed paraffin-embedded (anaplastic astrocytoma). Tissues were carefully washed with pre-cold PBS, and then minced for homogenization with tissue lysis buffer (Invitrogen). Written informed consent was obtained from each participant.
Cell viability, death, and apoptosis evaluations
Methyl thiazolyl tetrazolium (MTT) assays were performed for cell viability assessment. After different treatments, 0.5 mg/mL MTT reagent was added into the culture medium and then incubated for 4 hours at 37°C. Purple formazan salt crystals were dissolved by adding the solubilization solution with 10% sodium dodecyl sulfate (SDS) and 0.01 M HCl. The absorption at 490 nm was measured using a multi-well plate reader.
For cell death assessment, trypan blue staining approach was applied. Attached cells were digested with trypsin/ethylene diamine tetraacetic acid (EDTA), suspended in 1X PBS, and mixed with 0.4% trypan blue dye (Sigma, St. Louis, MO, USA). Dead cells would take up trypan blue due to compromised cell membranes, while viable cells would not due to the membrane integrity. The percentages of dead cell were recorded for further assessments. Cell Apoptosis DNA ELISA Detection Kit (Roche, San Francisco, CA, USA) was used for apoptosis assessment according to manufacturer’s instructions.
Cell proliferation, migration, and invasion evaluations
To evaluate cell proliferation, colony formation assays were performed. A172 cells were harvested with trypsin/EDTA and re-suspended in pre-cold PBS before being seeded in a 6-well plate at a density of 500 cells per well with completed DMEM medium (10% FBS). The cells were cultured for 21 days before the cell colonies were stained with crystal violet solution (Sigma). Number of viable colonies was recorded.
EID3, and AMPKα1 knocking down
Control lentivirus pLKO.1 vector and pLKO.1 vectors containing shRNA for EID3 (RHS4531-EG493861) were purchased from Thermo Fisher Scientific (Waltham, MA, USA). Glioma cells were infected with viral particles twice for the establishment of stable EID3 knocking down clones. Stable transfected cells were selected with puromycin (2 µg/mL). Knocking down efficiency were confirmed by real-time polymerase chain reaction (RT-PCR) and Western blot (WB) assays.
Two lentiviral AMPKα1 shRNAs were purchased from Santa Cruz Biotech (shAMPKα1-1; Santa Cruz, CA, USA) and GeneChem (shAMPKα1-2; Montreal, Canada). Each shRNA was added into A172 cells separately for 24 hours. The cell culture medium was then replaced by fresh medium for an additional 24 hours. Stable clones expressing AMPKα shRNA were selected by puromycin (0.5 µg/mL; Sigma) for a period of 10 days. Control cells were infected with lentiviral scramble shRNA (Santa Cruz Biotech). Knocking down efficiencies were evaluated by RT-PCR and WB assays.
Construction of AMPKα1 dominant negative mutation
The dominant negative AMPKα (DN-AMPKα, T172A) construct was designed as mentioned before. Glioma cells were cultured in a medium without antibiotics when reached 60–70% confluence. Then DN-AMPKα cDNA (0.10 µg/mL) was transfected into the glioma cells followed by Lipofectamine 2000 protocol. Afterwards, stable cells were selected via neomycin (1.0 µg/mL; Sigma). Transfection efficiency was verified via WB.
RT-PCR assays
Total RNA was extracted from cells using RNeasy Mini Kit (Qiagen, Dusseldorf, Germany), and reversed transcript using Quantitect Reverse Transcription Kit (Qiagen) according to the manufacturer’s instructions. The RT-PCR mixtures contained 2× QuantiTect SYBR Green PCR Master Mix (Qiagen), 10× QuantiTect primer assay mix, and synthesized cDNA. Amplification was performed in triplicate for each sample on a Lightcyle 480 platform (Roche, Mannheim, Germany). EID3 protein expression levels were normalized to glyceraldehyde-3-phosphate dehydrogenase (GAPDH) levels. Primer sequences were shown as follows : GADPH, forward (F), TGCACCACCAACTGCTTAGC; reverse (R), GGCATGGACTGTGGTCATGAG; EID3, F, GGAGGCACCTCAGTCATGATA; R, GTCCACAAACAGTTCTCTGAC.
WB assays
Cells were lysed in RIPA lysis buffer (Beyotime, Shanghai, China) with PMSF (Roche, Mannheim, Germany). Supernatant was collected after centrifugation at 13000 ×g at 4°C for 10 minutes. Protein concentration was measured with a BCA protein kit (Beyotime) and the whole lysates were mixed with 4× SDS loading buffer (125 mmol/Tris-HCl, 4% SDS, 20% glycerol, 100 mmol/L DTT, and 0.2% bromophenol blue). Protein samples were heated to 100°C for 5 minutes. For the WB, 30 µg total protein samples were loaded to a SDS-polyacrylamide gel. After electrophoresis, the protein was transferred to a polyvinylidene fluoride membrane blocked in 5% fat-free milk and incubated with antibodies against EID3 (1 : 800; Abcam, Burlingame, CA, USA), AMPKα1 (1 : 1000; CST, Chicago, IL, USA), pS6K1 (1 : 1000; Abcam), S6K1 (1 : 750; Abcam), LC3B-I (1 : 1000; Abcam), LC3B-II (1 : 800; Abcam), Becllin (1 : 750; Pharmingen, San Diego, CA, USA), ATG-5 (1 : 800; Abcam), ATG-7 (1 : 1000; Abcam), p62 (1 : 1000; Abcam), pAMPKα1 (1 : 1000; Abcam), ACC (1 : 1000; Abcam), pACC (1 : 1000; Abcam), and Tubulin (1 : 1000, Proteintech, Wuhan, China) overnight at 4°C, followed by another 2 hours incubation with horseradish peroxidase-conjugated anti-rabbit secondary antibody (1 : 5000; Jackson ImmunoResearch Labs, West Grove, PA, USA). GAPDH was served as a loading control. NIH Image J software (Bethesda, MD, USA) was used for the densitometric analysis of the WB. The protein levels were normalized with GAPDH.
Glioma Xenograft assays
Specific pathogen-free BALB/c nude mice (4–6 weeks old) purchased from the Chinese Academy of Medical Science were injected with 5×106 glioma cells subcutaneously into the left upper flank regions. The subcutaneous tumor size was calculated and recorded every week with vernier caliper as follows : tumor volume (mm3)=(L×W2)/2, where L=long axis and W=short axis, the measurements were repeated three times.
Construction of patients-derived xenografts models and primary glioma cells
Glioma patient-derived xenograft (PDX) models were generated according to a previous study [17]. Briefly, 4 weeks old male non-obese diabetic severe combined immune deficiency (NOD/SCID) mice were used as transplant recipient models and raised in the aseptic environment. Fresh glioma samples were cut into 1mm pieces within 1 hour after removal from patients. Tissue fragments were incubated in DMEM medium mixed with 50% Matrigel™ (BD, Franklin Lakes, NJ, USA), 10 ng/mL epidermal growth factor (Gibco), 10 ng/mL basic fibroblast growth factor (Gibco), 100 U/mL penicillin, and 100 U/mL streptomycin for 30 minutes. The whole tumor tissue mixture was then transplanted into the right flanks of mice (n=3; 4–5 weeks old; Shanghai Institute of Material Medicine, Chinese Academy of Science) subcutaneously with a #20 trocar. Animal welfare and experimental protocols were approved by the Shanghai Medical Experimental Animal Care Commission. Primary EID3 expression levels were detected by RT-PCR and WB prior to tissue transplantation. Single glioma cell suspension was made according to a previous study [6]. All the samples were mechanically disaggregated and digested with type IV collagenase (Gibco) within one hour, and resuspended in DMEM medium. The treated tissue was filtrated through a 40 µm filter to obtain the single-cell suspension. Red blood cells were lysed with ACK buffer (Invitrogen). The number of viable cells was counted and analyzed by trypan blue analysis (Procell, Wuhan, China).
Statistical analysis
Statistical analyses were performed with SPSS ver. 20.0 for Mac (IBM, Armonk, NY, USA). Quantitative data presented as mean±standard error are from at least three independent experiments. Continuous data were analyzed by one-way analysis of variance and Student’s t-test, and categorical data were analyzed by Fisher’s exact test or chi-square test. A p-value <0.05 was considered statistically significant.
RESULTS
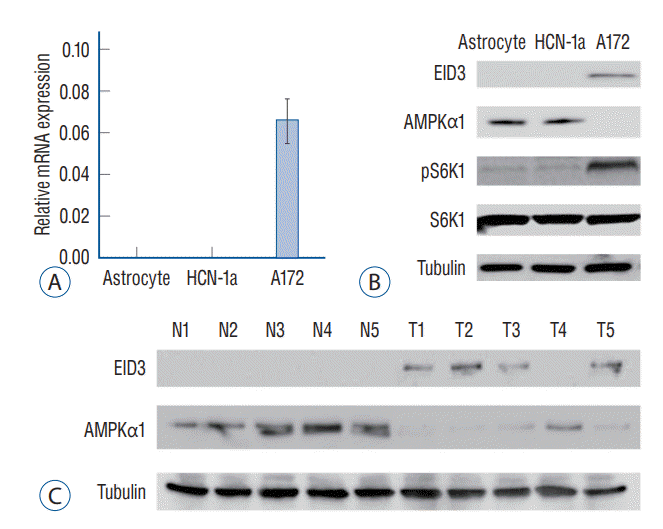
EID3 is preferentially expressed in glioma cells and correlated with AMPKα1
EID3 expression level in glioma cells was evaluated by quantitative PCR and WB. Results of RT-PCR assays showed that EID3 only expressed in glioma cells, and undetectable in human astrocyte and neuronal cells (HCN-1a cell line) (Fig. 1A). WB assays further validated the findings of RT-PCR (Fig. 1B). Since AMPK was identified as a key suppressor in glioma, we next investigate the correlation between EID3 and AMPKα1, an active form of AMPK. The WB results showed that EID3 expression was correlated with AMPKα1 downregulation (Fig. 1B). AMPK inhibition could result in mTORC1 activation process, during which phosphorylation of S6K1 was considered as a hallmark. Hence, we further explore the S6K1 activation status. Our data showed that the S6K1 phosphorylation was increased in AMPKα1-low A172 cells. However, the pS6K1 level was low in EID3-null astrocytes and HCN-1a cells which with a high AMPKα1 expression (Fig. 1B).
EID3 and AMPKα1 expression status were also evaluated in human glioma tissues. As shown in Fig. 1C, EID3 expression could be detected in four of five patients (80%) enrolled, however, it could not be detected in surrounding normal brain tissues. Also, EID3 expression was correlated with downregulation of AMPKα1, which consistent with the results in A172 cells.
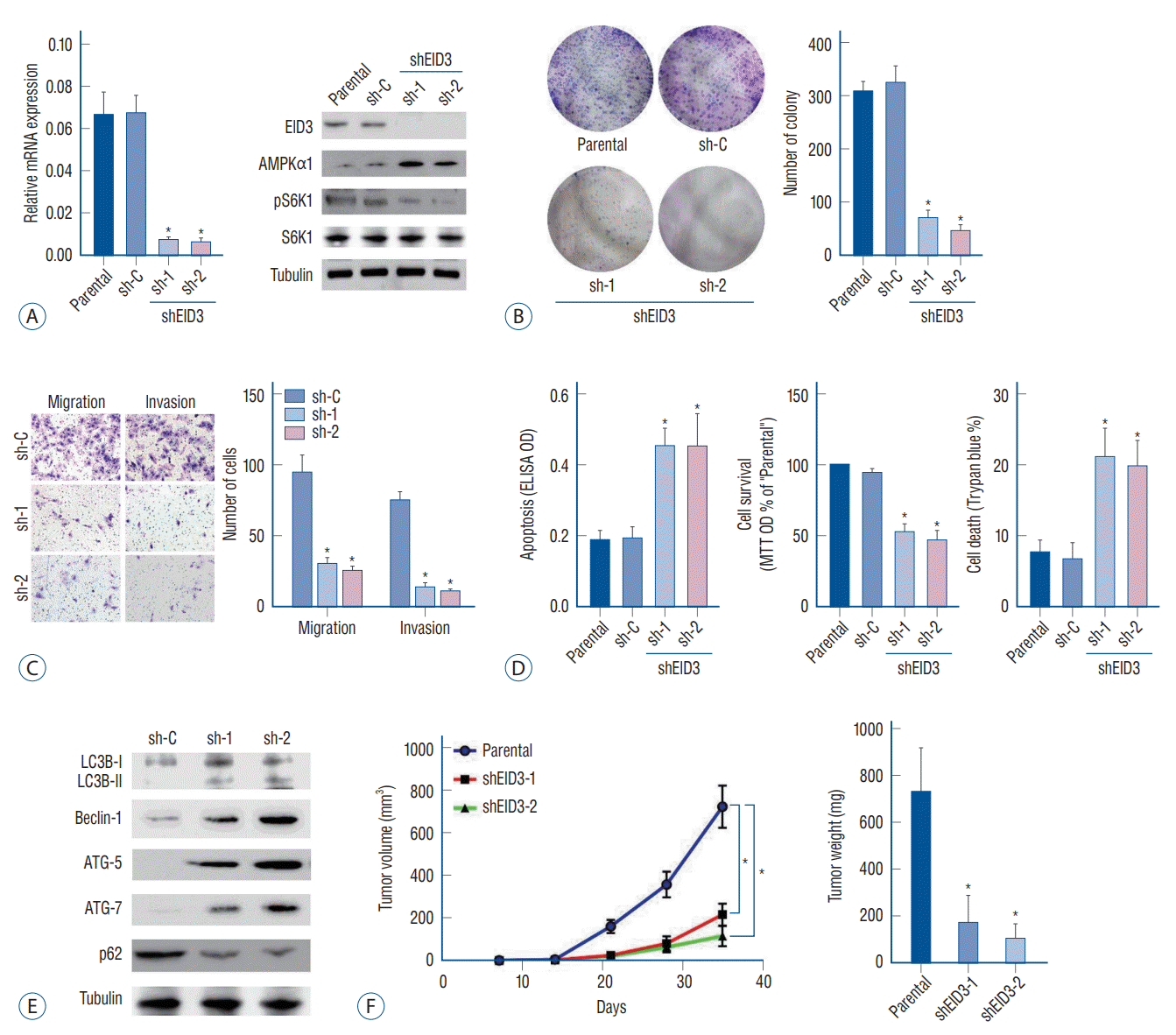
EID3 knocking down inhibited proliferation, survival, and invasion of glioma cell
EID3 expression was knocking down with two shRNAs targeting different sites of EID3 cDNA, the efficiencies of knocking down were validated through RT-PCR and WB (Fig. 2A). Remarkably, EID3 knocking down could restore AMPKα1 expression, resulting in inhibition of S6K1 (Fig. 2A). Colony formation assays demonstrated that EID3 knocking down significantly inhibited A172 cell proliferation (p<0.05, Fig. 2B). Meanwhile, Transwell assays indicated that the number of migrated and invaded cells was significantly decreased when EID3 expression is knocked down (Fig. 2C and Supplementary Fig. 1). Based on the MTT results, we found the number of viable cells was significantly decreased after EID3 knocking down. We also found that EID3 shRNA could induce cell death according to trypan blue staining. Additionally, Histone DNA apoptosis ELISA assay revealed EID3 shRNA could induce apoptosis in glioma cells (Fig. 2D), while the scramble non-sense control shRNA exhibited no effects on glioma cell proliferation, death, and apoptosis.
According to a previous report [22], AMPK activation would trigger autophagy by activating Ulk1 or inhibiting mTOR signaling. Since our results indicated that EID3 could restore mTOR activation by downregulating AMPKα1, we further investigated the effects of EID3 on autophagy in glioma cells. Our data indicated that EID3 shRNA could induce increased Beclin-1, ATG-5, and ATG-7 expression, LC3B-I to LC3B-II switch, and p62 degradation, which confirmed that EID3 knocking down glioma cells exhibited autophagy-like changes (Fig. 2E).
We also validated in vivo proliferation inhibition of EID3. A172 cells that stable express EID3 shRNAs and parental control A172 cells were injected into the flanks of SCID mice, and tumor volumes and weights were recorded. As Fig. 2F demonstrated, tumors generated from EID3 knocking down A172 cells grew significantly slower than control tumors (sh-C). Moreover, weights of tumor generated from EID3 knocking down A172 cells were significantly lighter than those of control tumors.
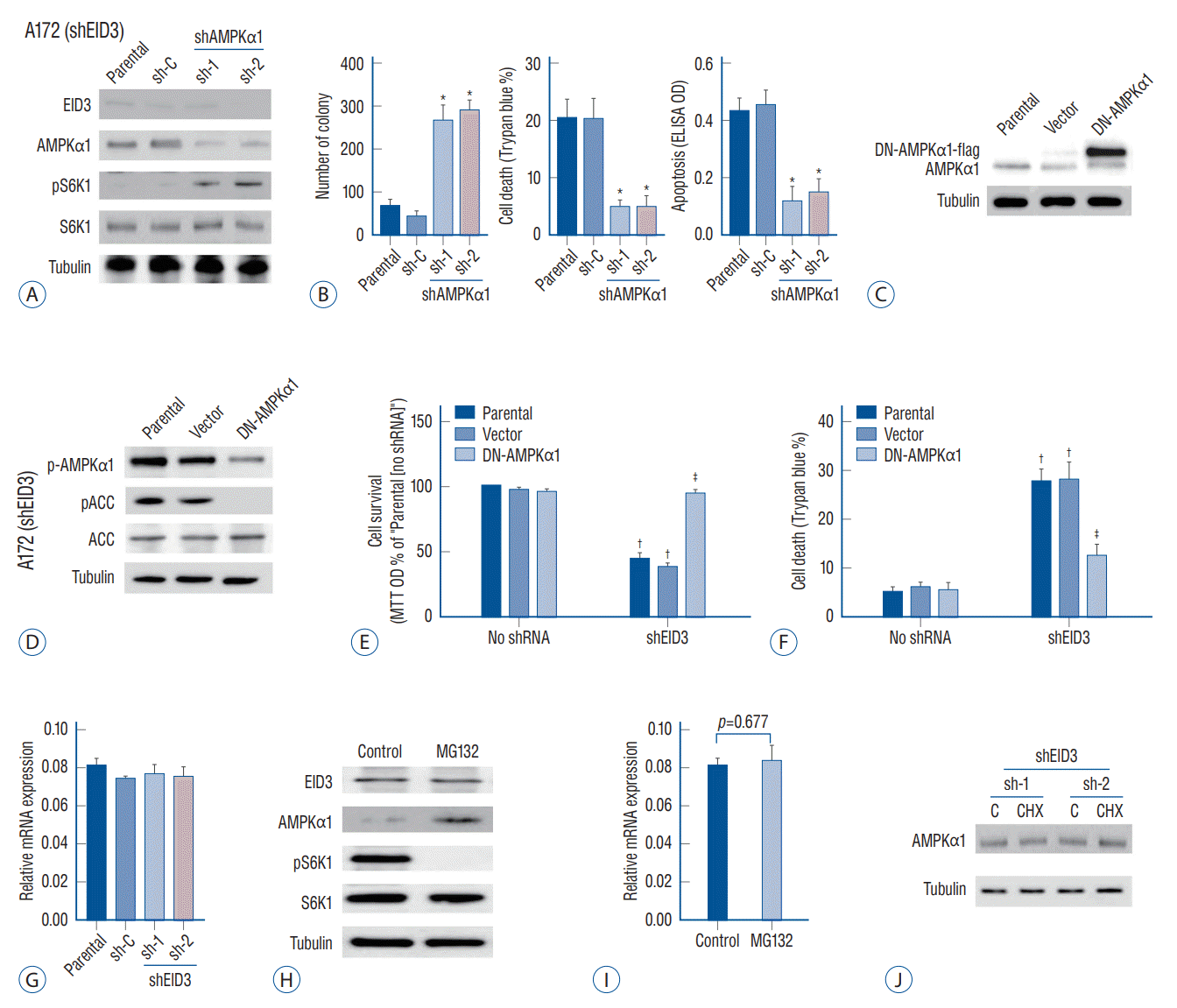
Validation of AMPKα1 as the key downstream molecule of EID3 pathway, EID3 might decrease AMPKα1 level by promoting protein degradation
Our results indicated that EID3 knocking down could restore AMPKα1 expression and inhibit tumor growth and invasion. We further explored whether AMPKα1 served as the key inhibitor that mediated the inhibition effects following EID3 knocking down. For this purpose, AMPKα1 was knocked down in A172 cells expressing shEID3 (Fig. 3A). We found that AMPKα1 inhibition could restore the activation of S6K1 in EID3 knocking down cells (Fig. 3A). However, AMPKα1 knocking down did not affect expression status of EID3. Moreover, cell viability reduction and cell apoptosis caused by EID3 knocking down could be largely attenuated by AMPKα1 knocking down inhibition (Fig. 3B).
To further study the role of AMPKα1, a dominant negative AMPKα1 mutant (DN-AMPKα1, T172A) were introduced into A172 cells, aiming to block AMPKα1 activation following EID3 knocking down. WB assays confirmed exogenous expression of the DN-AMPKα1 in A172 cells (Fig. 3C). As expected, EID3 knocking down inhibition greatly activated AMPKα1, which can be proven by increased expression levels of p-AMPKα1 and p-ACC (Fig. 3D). However, such effects were significantly hindered by DN-AMPKα1 (Fig. 3D). Consistently, forced expression of DN-AMPKα1 did not rescue the inhibition effects of EID3 knocking down on cell survival (Fig. 3E) and death (Fig. 3F). Meanwhile, all control shRNAs showed no significant effects on A172 cells. Through RT-PCR assays, we found that neither EID3 shRNA could decrease AMPKα1 mRNA expression (Fig. 3G), which suggested EID3 regulates AMPKα1 expression through a non-transcriptional way. Therefore, MG132, a proteasome inhibitor, was introduced to test our hypothesis. WB assays showed that MG132 restored AMPKα1 expression in A172 cells with high EID3 expression (Fig. 3H). Also, MG132 reactivated the mTOR signaling, supported by increased pS6K1 level after treatment (Fig. 3H). However, MG132 showed no significant influence on the mRNA expression of AMPKα1 (Fig. 3I). To exclude the possibility that EID3 inhibits the protein synthesis process of AMPKα1, cycloheeximide (CHX), an inhibitor of protein synthesis was used. Results showed that CHX treatment could not affect the expression level of AMPKα1 in EID3 knocking down A172 cells (Fig. 3J), which indicated that increased expression of AMPKα1 in EID3 knocking down cells might not due to promoted protein synthesis. Collectively, these data implied that EID3 might induce ubiquitination and proteasomal degradation of AMPKα1.
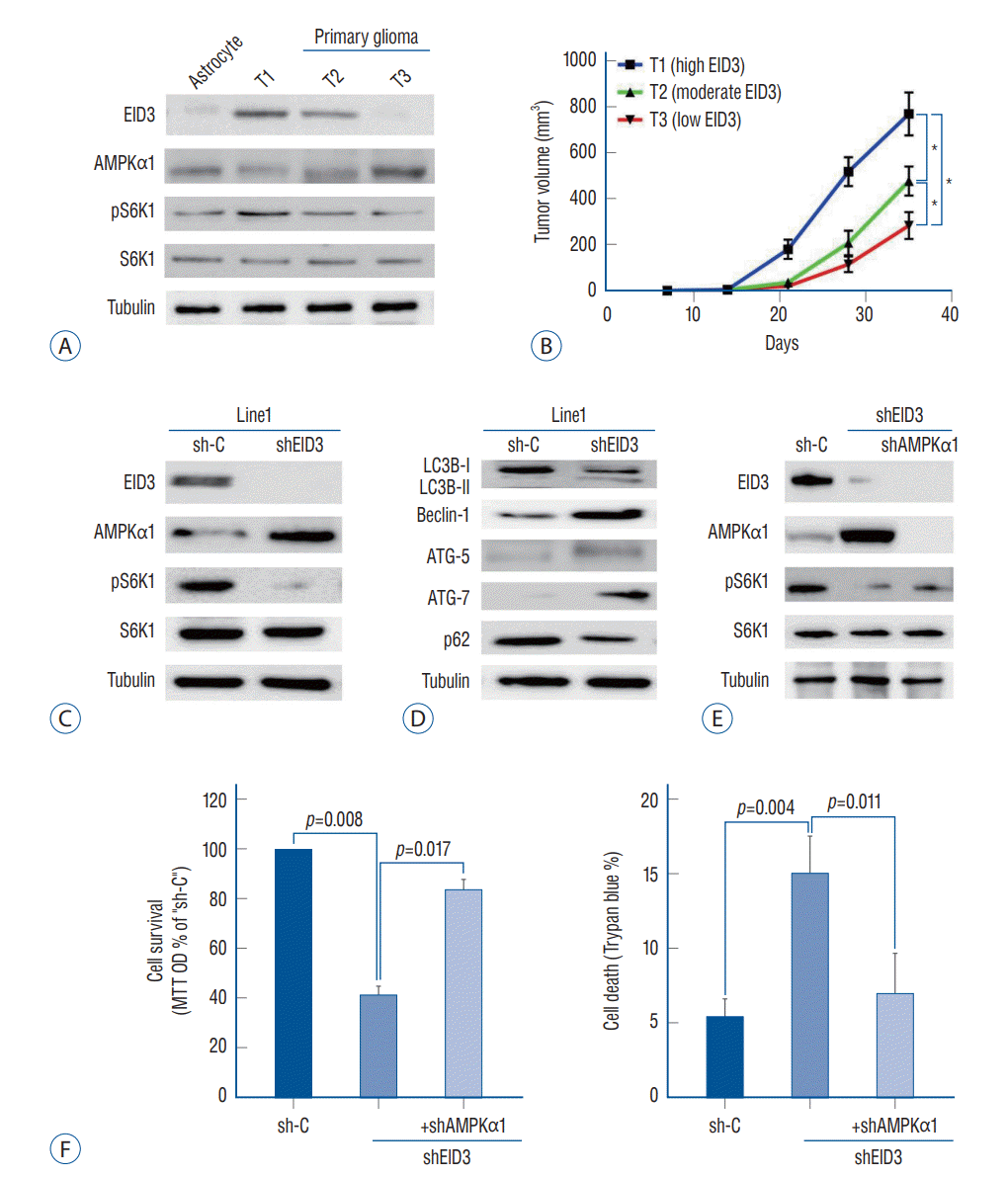
AMPK mediated viability reduction and cell death in EID3 knocking down primary glioma cells
To validate the function of EID3 in primary glioma, we generated three patient-derived xenograft model with descending EID3 expression (T1, high; T2, moderate; T3, low). The EID3 expression status were confirmed by WB assays (Fig. 4A). Accordingly, primary glioma with high EID3 expression showed lower level of AMPKα1 but high pS6K1 expression (T1), while tumor with undetectable EID3 exhibited high level of AMPKα1 and low level pS6K1 (T3, Fig. 4A). We observed that T1 (high EID3) grew significantly faster than T2 and T3, based on bigger tumor volumes (Fig. 4B). Moreover, T2 also showed significantly faster growth than T3 did (Fig. 4B).
We then separated single glioma cells from T1 tumor tissue and transfected it with EID3 shRNA to explore the effects of EID3 knocking down in primary glioma. WB assays confirmed the efficiencies of EID3 knocking down and demonstrated that EID3 knocking down resulted in upregulation of AMPKα1 and inactivation of S6K1 (Fig. 4C). Furthermore, we also observed an autophagy-like phenotype in primary glioma cells with EID3 knocking down, which were similar to those findings in A172 cells (Fig. 4D). To further validate the role of AMPKα1, primary T1 glioma cells were transfected with AMPKα1 shRNA. Notably, AMPKα1 knocking down could effectively restore the pS6K1 level in primary glioma cells transfected with EID3 shRNAs (Fig. 4E). Moreover, AMPKα1 knocking down largely attenuated the viability reduction and cell death caused by EID3 inhibition (Fig. 4F). These results in primary glioma cells further confirmed that AMPK mediated viability reduction and cell death by EID3 knocking down.
DISCUSSION
Present study identified EID3 as a novel oncogenic molecule in human glioma, and was critical for glioma cell survival, proliferation and invasion. EID3 was preferentially expressed in glioma tissues/cells, while undetectable in astrocytes, neuronal cells, or normal brain tissues. Importantly, EID3 knocking down significantly inhibited in vivo tumor growth in SCID mice. Moreover, we found EID3 expression positively correlated with tumor growth in PDX models. AMPKα1 was identified as the key downstream regulator underlying EID3 that mediated its suppression effects when being knocked down, and EID3 might downregulate AMPKα1 through protein degradation.
EID3 was identified as a negative regulator of cellular differentiation, as it binding to class I HDAC or CBP/p300 [15,27]. However, its function in human cancer, especially in glioma, remained largely unclear. A pilot study revealed that EID3 might be a critical promoter for the carcinogenesis and development of colorectal cancer [27], which led to our hypothesis that EID3 might also serve as an oncogene in glioma. Our results showed that EID3 knocking down significantly hindered glioma cell proliferation and invasion, induced cell viability reduction, apoptosis, and cell death. Therefore, we believed EID3 acted as a vital regulator in glioma that promoting tumor progression. This characteristic and novel function of EID3 was shown for the first time in human glioma.
AMPK has been considered as a critical tumor suppressor for a long time, and AMPK signaling has been reported to be dysregulated in various kinds of solid tumors [20]. In lung and cervical cancer, AMPK signaling was inhibited by loss-off-unction or deletion of upstream activator [13,29]. In hepatocellular carcinoma (HCC), AMPK signaling was inhibited by insulin growth factor pathway activation, and metformin showed anti-HCC effect via reactivating AMPK signaling [35,37]. In glioma, AMPK signaling was also constantly inhibited, and several upstream regulators, such as MAGEA6, have been identified [30]. In present study, we showed that EID3 served as a novel AMPK signaling suppressor, since EID3 expressed glioma tissues/cells showed decreased expression of AMPKα1. Meanwhile, AMPKα1 knocking down could effectively rescue glioma cells from apoptosis and death caused by EID3 silence. However, we only figured out that EID3 might downregulate AMPKα1 expression through proteasomal degradation, while detailed molecular mechanism involved in this process remained unclear. Further investigation is needed to explore EID3 induced AMPKα1 degradation and key molecules involved in this process.
mTOR hyper-activation was considered as a hallmark in glioma, which greatly contributed to carcinogenesis [33]. Therefore, great efforts have been made to develop novel regimens targeting mTOR or it regulated signaling pathways. Previous study showed that AMPK activation could suppress mTOR signaling through various ways, such as phosphorylation of TSC2, which was a key upstream inhibitor of mTOR signaling [34]. AMPK could also phosphorylate and inactivate raptor to block mTOR signaling activation [12]. Restoration of AMPK signaling might be an ideal strategy for preventing glioma progression. In present study, we showed that AMPK expression recovered from EID3 silencing, and recovery of AMPK could effectively inhibited mTOR activation, resulting in viability reduction, cell death and apoptosis. Significantly, mTOR signaling could be restored in EID3 knocking down cells through transfecting AMPKα1 shRNA or plasmid containing dominant negative AMPKα1 mutation. Our data implied that AMPK activation caused by EID3 silence was responsible for the inhibition of mTOR signaling in glioma.
CONCLUSION
Taken together, our study demonstrated that EID3 promoted glioma cell proliferation and survival through inducing AMPKα1 downregulation. These findings strongly implied EID3 as a novel biomarker and target for glioma. Notably, targeting EID3 might represent a promising strategy for treating mTOR in glioma. Further investigations aiming to screen and identify selective inhibitor EID3 are urgently needed.
 XML Download
XML Download