

 PDF
PDF Citation
Citation Print
Print



INTRODUCTION
Traumatic brain injury (TBI) primarily causes structural damages to the brain, resulting in decreased consciousness, disrupted blood-brain barrier (BBB), and cerebral edema. In addition to the primary brain injury, it can also affect other end organs, especially the lungs, and can worsen the patient’s prognosis if the complications in the end organs are not properly treated [10]. Rincon et al. [15] reported that the prevalence rate of acute respiratory distress syndrome (ARDS) and acute lung injury (ALI) was 22% and the rate of ARDS/ALI-related mortality was 28% in patients with TBI. TBI patients are at risk of developing pulmonary complications from the accompanying chest trauma. However, pulmonary complications can also occur due to the systemic inflammatory mediators released due to TBI [13]. Weber et al. [18] reported that receptor for advanced glycation end products (RAGE)-sufficient mice showed systemic hypoxia and ALI compared to RAGE-deficient mice. In addition, neutralizing systemic high-mobility group box-1 improved hypoxia and lung compliance. Kerr et al. [10] also reported that extracellular vesicles mediated the activation of pulmonary inflammation after TBI. These data suggest that TBI-induced pulmonary complications are common and associated with activation of a neural-respiratory-inflammatory axis.
It is widely believed that acute pulmonary complications occurring after TBI are usually accompanied by severe TBI. Acute severe TBI contributes to a rapid sympathetic surge and results in pulmonary edema by increased systemic vascular resistance [4]. However, mild TBI can also cause pulmonary complications. Humphries et al. [8] reported that mild TBI induced pulmonary neutrophil priming, and thus, subsequent insults such as acid aspiration can cause massive alveolar inflammation. Theoretically, the systematic response of innate immunity activation is highly likely to be involved in acute pulmonary complications after mild TBI. Among the various factors involved in innate immunity, many studies have focused on neutrophils in the pulmonary response after TBI [17]. However, other cellular components of innate immunity, in particular mast cells, may also be crucial in the systematic response to TBI [5]. Here, we evaluated the histological changes and fluctuations in inflammatory markers in the lungs to determine whether mild TBI induced an acute pulmonary inflammatory response. In addition, we focused on mast cells to determine their role in pulmonary inflammation.
MATERIALS AND METHODS
In vivo TBI model and tissue preparation
All animal experiments were approved by the Institutional Animal Care and Use Committee (IACUC) of Hallym University (HallymR2 2020-51). All experimental mice (n=48) were randomly divided into sham-operated (n=24) and experimental mild TBI (n=24) groups. In addition, the two groups were subdivided into three groups measured 6 hours, 24 hours, and 72 hours after trauma (Fig. 1A and Supplementary Fig. 1). Then, the mice were anesthetized and euthanized immediately thereafter.
In vivo TBI was induced via open-head injury using a stereotaxic impactor (RWD Life Science, Shenzhen, China) [20]. In brief, the mice were anesthetized using 2.5% isoflurane in oxygen and placed in the stereotaxic frame. The skull was exposed via a midline skin incision, and TBI was induced as follows : M/L -2.5 mm and A/P -2.0 mm from the bregma at a depth of 1.5 mm using a blunt tip with a diameter of 2 mm. The velocity of the impactor reached 3.0 m/s with a depth of 1.5 mm using the 2-mm blunt tip below the dura matter. The dwell time in the brain was 0.5 m/s (Fig. 1B and C).
Brain and lung tissues were harvested and transcardially perfused with 4% paraformaldehyde. Brain tissues were removed and fixed for 24 hours in paraformaldehyde, at which time they were cryo-protected in 20% sucrose for 48 hours. The brain tissues were blocked with optimal cutting temperature compound and stored at -80℃ until serially cut into 10-µm coronal sections. The lung tissues were embedded in paraffin blocks and sectioned into 5-µm thick slices and stored at room temperature.
Fluoro-Jade B (FJB) staining
The brain tissue slides were hydrated in distilled water for 1 minute and immersed in 0.06% potassium permanganate for 15 minutes at room temperature. The slides were stained with 0.001% FJB (Histo-Chem Inc., Jefferson, AR, USA) solution for 30 minutes. After washing the stained slides three times in distilled water for 5 minutes, the slides were dried at 55℃ in the dark for at least 35 minutes and mounted with dibutyl phthalate polystyrene xylene (Sigma-Aldrich Co., Saint Louis, MO, USA). FJB-stained brain tissue images were obtained with light microscopy (Carl Zeiss, Oberkochen, Germany) at ×20 magnification. The stained brain tissues were observed at 450–490 nm using a fluorescence microscope.
Hematoxylin and Eosin (H&E) staining
Slides of paraffin-sectioned lung tissue were incubated in a tissue-drying oven for 30 minutes at 60℃ and then stained with H&E according to IHC-WORLD protocols (http://www.ihcworld.com/_protocols
). H&E-stained lung tissue images were obtained by light microscopy at ×20 and ×40 magnifications. Mast cell counts were performed on Astra blue-stained preparations under oil immersion microscopy and obtained at ×100 magnification. Quantification of the mast cells distributed in the lungs was performed and analyzed using ImageJ software (version 1.49v; National Institutes of Health, Bethesda, MD, USA).
RNA isolation and quantitative real-time polymerase chain reaction (qRT-PCR) analysis
Total RNA was extracted using the easy-BLUE Total RNA kit and reverse transcribed into cDNA using the Maxime RT PreMix Kit (iNtRON Biotechnology, Inc., Burlington, MA, USA). qRT-PCR was performed in triplicate for each sample using the SYBR Green PCR Kit (applied Biosystems, Foster, CA, USA) for 40 cycles with a 3-step program including 15 seconds of denaturation at 94℃, 30 seconds of annealing at 55℃, and 30 secons of extension at 70℃. Amplification specificity was assessed by melting curve analysis. The sequences of the qRT-PCR primers are presented in Supplementary Table 1. Glyceraldehyde-3-phosphate dehydrogenase was used as the endogenous control. The expression in qRT-PCR was analyzed using the 2-ΔΔct method.
Statistical analysis
All data are presented as the median and range. The two-tailed non-parametric Mann-Whitney U test was used for the comparisons of independent samples [3]. A p-value of less than <0.05 is represented by an asterisk (*) in the Figs. 2 and 3 [2]. All statistical analyses were performed using GraphPad Prism software (v.6.01; GraphPad Software Inc., San Diego, CA, USA).
RESULTS
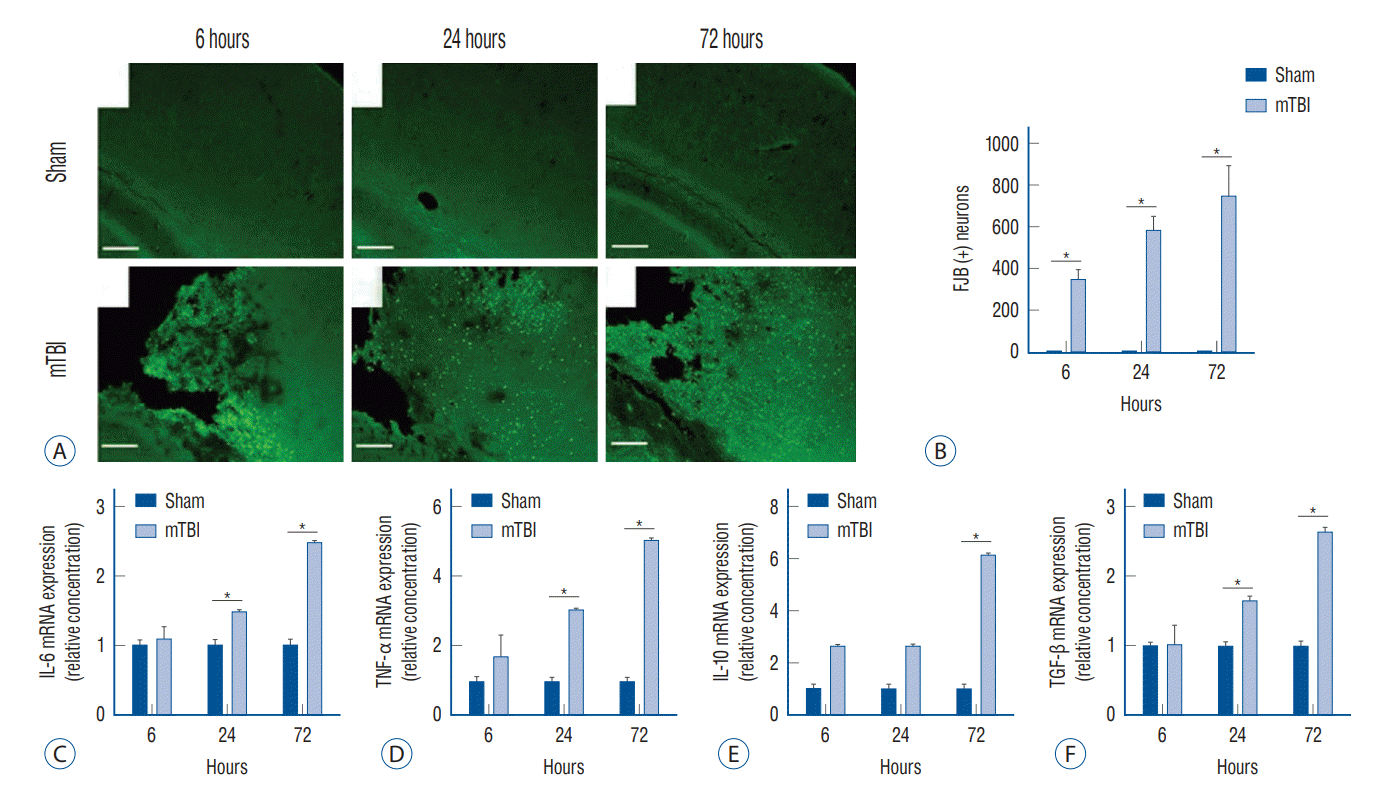
Neuronal degeneration and inflammation after mild TBI
We evaluated and compared neuronal degeneration and the inflammatory cytokines in the brain over time following mild TBI. Compared to the sham-operated group, mice with mild TBI had significantly increased neuronal degeneration, shown as FJB-positivity, at all measurement points (Fig. 2A and B). In addition, the degree of degenerative changes showed a tendency to increase with time. Mild TBI increased the mRNA expression of interleukin (IL)-6, tumor necrosis factor (TNF)-α, IL-10, and transforming growth factor (TGF)-β in the injured brains (Fig. 2C-F). In particular, the expression of IL-6, TNF-α, and TGF-β was significantly increased in mild TBI 24 hours after injury compared to the sham-operated mice, and this was more pronounced at 72 hours.
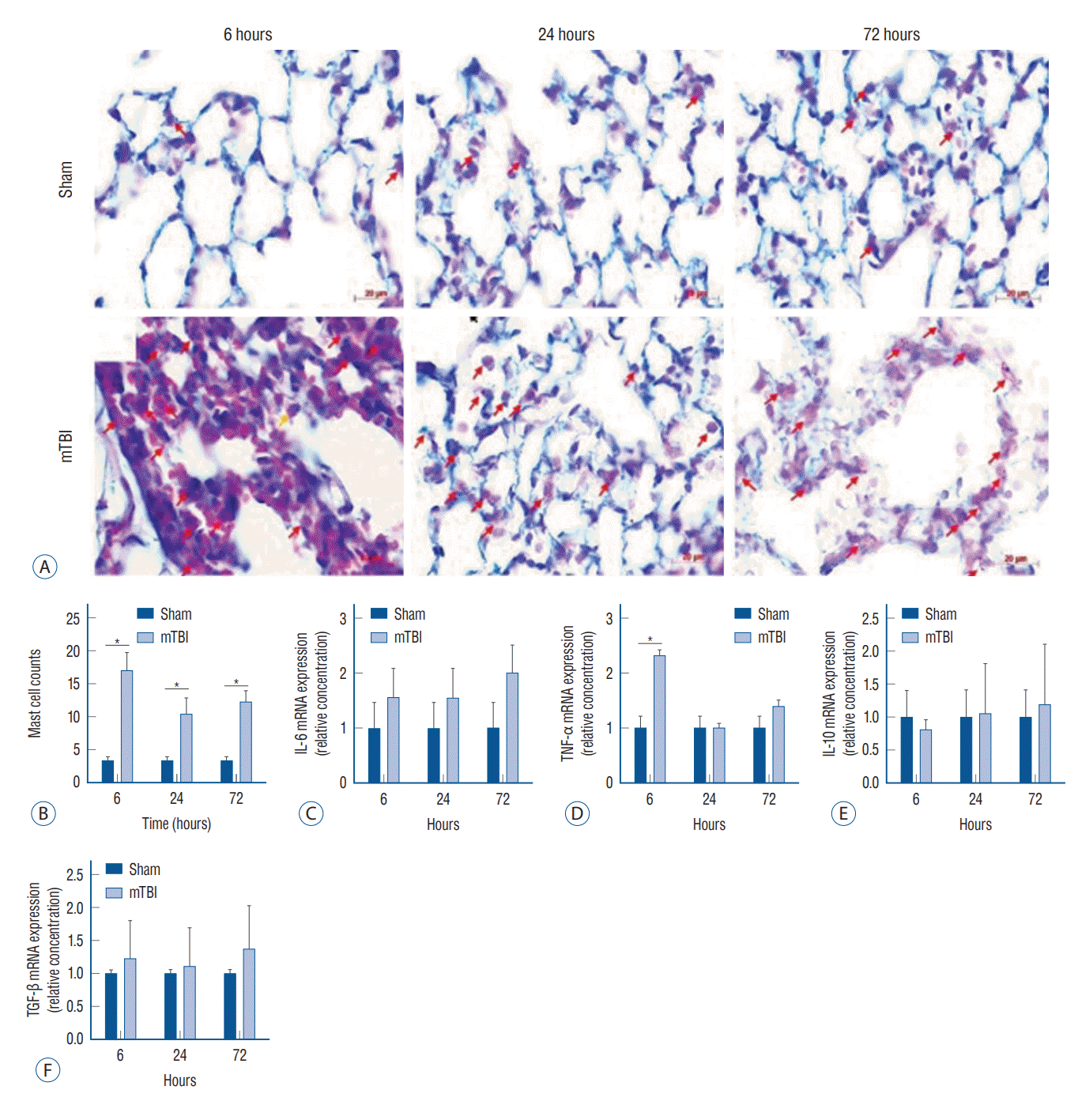
Acute pulmonary inflammatory response after mild TBI
Staining of lung tissues using H&E was performed to clarify the morphological changes after TBI. Overall, the pulmonary histological architecture after mild TBI presented as interstitial edema with cell infiltration, which was particularly pronounced 6 hours after the insult (Supplementary Fig. 2). As time elapsed, interstitial cell infiltration decreased, but the morphological changes of alveolar and partial disruption of the capillary membranes were observed. To clarify the type of infiltrated cells more precisely, Astra blue and hematoxylin staining was performed (Fig. 3). Six hours after mild TBI, a significant infiltration of mast cells was observed compared to the sham-operated mice. In the lungs, the mRNA expression of TNF-α was significantly increased 6 hours after mild TBI, but there was no significant difference 24 and 72 hours after the insults. Also, there was no significant difference in other inflammatory cytokines compared to the sham-operated mice.
DISCUSSION
TBI can induce pulmonary damage by increasing immunoreactivity in the lungs via a neural-respiratory inflammatory axis. In general, there is a tendency to think that the neural-respiratory inflammatory axis is mainly involved following moderate-to-severe TBI. Endogenous danger-associated molecular pattern protein is released into the extracellular space after TBI and associated with local pulmonary inflammation by binding to the receptors. However, focal injury in the brain parenchyma can also influence lung conditions. Austin et al. [1] reported that increased numbers of macrophages were observed in bronchoalveolar fluid and the increased mRNA expression of IL-1β was seen in whole lung tissues after focal ischemic stroke. After concussion by fluid percussion, neutrophil accumulation was observed in the lung interstitium. In this circumstance, even a small stimulus, defined as acid microaspiration in the study, could cause alveolar neutrophil influx and ALI [8]. Vaickus et al. [17] examined substance P and the neurokinin 1 receptor (NK1R) in mice lungs after mild TBI or blunt tail trauma. Compared to tail trauma, mild TBI induced increased expression of NK1R and enhance neutrophil to Pseudomonas aeruginosa infection. Our study also demonstrated that acute inflammation in the lung could occur in response to systematic inflammation caused by mild TBI.
We think that this inflammatory response can trigger an inflammatory cascade by activating the surrounding tissues, and resulting in inflammatory cells in the lung, particualrly mast cells. Excessive systemic inflammation by the activation of innate immunity has been suggested to be related to the morbidity and mortality of trauma patients [14]. Among the components of the innate immune systems, the role of mast cells has not been well-studied after mild TBI compared to neutrophils. Mast cells are spread all over the body [9] and mast cells present in the brain are highly involved in inflammation that occurs after brain injury and are associated with neurodegeneration. Mast cells continuously secrete inflammatory mediators through interactions with microglia and astrocytes and increase BBB permeability and leakage, leading to neurological complications [9]. Cai et al. [5] reported that mast cell-deficient mice had significantly decreased systematic inflammation and end-organ injuries and better hemodynamic stability than wild-type mice. Wild-type mice showed end-organ damage with increased alanine transaminase, aspartate aminotransferase, and tissue necrosis. In our study, TNF-α was significantly increased in the lungs 6 hours after TBI, and mast cell infiltration was clearly observed in the pulmonary interstitium. After bacterial infection, the mast-cell chemoattractant TNF-α was involved in the neutrophil response. Moreover, TNF-α-targeted antibodies decreased over 70% of the neutrophil influx [12]. Surely, the mechanisms of bacterial pulmonary infection and TBI-induced pulmonary injury are different, but we think that the TNF-α release by mast cells due to the initial host systemic immune response to mild TBI may involve an acute pulmonary inflammatory response.
Unlike previous studies, our findings illustrated the importance of the following aspects. First, we examined the pulmonary inflammatory response for up to 72 hours, focusing on mast cells after mild TBI. Although pulmonary interstitial edema was not pronounced 72 hours after mild TBI, mast cell invasion into alveolar cells and alveolar structural changes were observed 72 hours after the insults. Since the patient’s prognosis can be persistently vulnerable to pulmonary infection due to alveolar structural changes, efforts to reduce mast cell-related pulmonary inflammation in the early stage after mild TBI are needed. Second, we measured inflammatory markers after mild TBI in brains and lungs. In the injured brain, the inflammatory cytokines gradually increased over time, showing a maximum difference compared to the shamoperated group at 72 hours, whereas in the lung, the greatest difference in TNF-α occurred 6 hours after the insults. We postulated that mast cells mediate inflammatory processes through TNF-α release in the pulmonary response to mild TBI. Therefore, variations in the degree of post-traumatic inflammation in the brain and lungs by period implied that the focus of treatment should be different for each period to improve the patient’s prognosis. Finally, we further analyzed whether there was a difference in mast cell counts 24 and 72 hours after TBI. Although the number of mast cells measured at the two different times did not differ significantly, there is a possibility that mast cells may increase as chronic inflammation progresses [7]. Accordingly, our findings mainly correspond to the role of mast cells in acute post-traumatic inflammation.
There were some limitations to our study. First, we did not analyze bronchoalveolar lavage fluid (BALF) after mild TBI. The cell count and type can reflect the degree of pulmonary injury as well as the response to treatment. The findings of this study suggest that mast cells play an important role in pulmonary inflammation after mild TBI. Accordingly, additional studies on the effect of mast cell inactivation should be done via BALF analysis. Second, there might be a discrepancy between the definition and the related severity of mild TBI in the mouse experimental model and actual clinical patients. Mild TBI patients are clinically defined by Glasgow coma scale scores between 13 and 15 with physiological disruptions such as a loss of consciousness of fewer than 30 minutes, post-traumatic amnesia of <24 hours, and transient neurological deficits [11]. There is debate as to whether the results of imaging tests should be included. However, in general, the absence of intracranial hemorrhage is often called mild TBI in clinical practice. Thus, our results may reflect a slightly more severe form than actual clinically mild TBI, although we performed a well-established and commonly used mild TBI model [19]. Third, several factors such as the anesthetic agent and microaspiration might influence the experimental results. The clinical efficacy of isoflurane in reducing lung damage is controversial, but mice treated with isoflurane exhibited significantly less physiologic lung dysfunction with decreased vascular leakage [6]. Microaspiration also might occur after TBI or during anesthesia. A multicenter study reported that 67% of the patients had abundant microaspirations of oral and gastric contents [16]. Therefore, it is necessary to evaluate the effect of isoflurane on TBI-associated lung injury through comparative analysis with other anesthetics and microaspiration after mild TBI and the resulting lung injury in future follow-up studies.
CONCLUSION
Mild TBI induced acute pulmonary inflammatory responses such as interstitial inflammation and alveolar structural changes. During this process, mast cells played an important role in pulmonary inflammation. Targeting mast cells in pulmonary complications after mild TBI may provide a novel therapeutic approach in neurocritical care medicine. Further studies on the clinical implications of the connection between mild TBI and acute pulmonary inflammation are needed.

 XML Download
XML Download