

 PDF
PDF Citation
Citation Print
Print



INTRODUCTION
Angiogenesis is a complex process in which blood vessels sprout from the existing microvasculature. Angiogenesis involves the coordination of many events, including disruption of the basement membrane (BM) by proteases, endothelial cell (EC) proliferation and migration, lumen formation, reassembly of the BM, recruitment of pericytes and/or vascular smooth muscle cells, vascular maturation, and, finally, new blood flow [1].
Under physiological conditions, angiogenesis is controlled by the balance among endogenous stimulators and inhibitors. Under pathological conditions (including tumor growth and chronic inflammation), the imbalance between stimulator and inhibitor levels triggers the so-called “angiogenic switch” [2]. Various pro-angiogenic factors are known, including vascular endothelial growth factor (VEGF) [3]. An increasing number of anti-angiogenic factors (including angiostatin and endostatin) have also been described [4]. VEGF is one of the most powerful angiogenic factors, directly affecting EC proliferation, migration, and tube formation under physiological and pathological conditions [5]. VEGF also plays an important role during the regulation of capillary actions and BM permeability of nasal polyps (NPs), leading to extensive edema and polyp growth [6].
Angiostatin exerts its anti-angiogenic effects by inducing EC apoptosis [7–9] and impairing EC proliferation [10], migration, and tube formation [11]. Endostatin induces EC apoptosis via various signaling pathways [12,13], and it suppresses EC migration, adhesion, and proliferation [14,15].
The role of angiostatin or endostatin in NP pathophysiology (angiogenesis) remains unclear. For this reason, we compared the accurate figure of angiostatin, endostatin, and VEGF in NPs and inferior turbinate mucosal tissues (control) to reveal it.
METHODS
Subjects
Thirteen patients with NPs and twelve patients having nasal septum deviation were enrolled at the department of Otorhinolaryngology-head & neck surgery, Inje University Busan Paik Hospital, Busan, Korea. NPs were diagnosed using the previously known criteria for chronic rhinosinusitis with NPs [16]. Written informed consent was obtained from all patients. Inje University Busan Paik Hospital Ethics Committee approved this study (IRB No. 16-0213). A NP was defined as visible polyp growing from the middle nasal meatus into the nasal cavity by endoscopy, affecting the ethmoid and maxillary sinuses as computed tomography of the paranasal sinus revealed. NPs were removed from the region of the middle meatus at the commencement of operation. Control inferior turbinate and nasal mucosal tissues were harvested from patients without evidence of NPs, to avoid any possibility to share chronic mucosal inflammation, who went through septoplasty with inferior turbinoplasty. No subject had any history of asthma, aspirin sensitivity, or nasal allergy. Allergies were diagnosed based on both a relevant history and the results of the Multiple Allergen Stimulation Test (MAST Immunosystems, Mountain View, CA, USA) and blood eosinophil count. No patient had taken systemic or topical steroids, antihistamines, macrolide antibiotics, or nonsteroidal anti-inflammatory drugs for 4 weeks before the procedure.
Reagents
3,3′-Diaminobenzidine (DAB) was purchased from Abcam Inc. (Cambridge, MA, USA). Antibodies against VEGF (cat. no. 250907) and endostatin (cat. no. 251198) were obtained from Abbiotec Inc. (San Diego, CA, USA). Antibodies against angiostatin (cat. no. bs-1828R) were purchased from Bioss Inc. (Woburn, MA, USA). Horseradish peroxidase (HRP)-conjugated anti-goat (SC-2020) antibodies were used as secondary antibodies (Santa Cruz Biotechnology, Inc., Dallas, TX, USA).
Protein extraction and Western blot analysis
Tissues were mortar-ground into fine powders in liquid nitrogen, transferred to 1.5-mL microfuge tubes, and lysis buffer (G-biosciences, St. Louis, MO, USA) was added. The proteins were separated on polyacrylamide mini-gels and transferred to nitrocellulose membranes (GE Healthcare Life Sciences, Little Chalfont, UK). Following overnight incubation with the appropriated primary antibody, the membranes were incubated with a secondary antibody conjugated to HRP. The immunoreactive bands were visualized using an electrochemiluminescence detection system (Pierce Biotechnology, Inc., Rockford, IL, USA).
Immunohistochemistry
Formalin-fixed paraffin-embedded NP and inferior turbinate mucosal samples were sectioned at a 5-μm thickness, mounted on poly-L-lysine coated slides, and deparaffinized and rehydrated via a series of xylene and alcohol. After rinsing with 1X phosphate-buffered saline, the sections were immersed in Target Retrieval Solution (pH 6.0; Dako, Carpinteria, CA, USA) for 5 min at 121°C in a pressure cooker and allowed to cool for 20 min. Endogenous peroxidase activity was blocked by incubation with Peroxidase-Blocking Solution (Dako) for 10 min at room temperature, and the slides were then washed in phosphate-buffered saline and immersed in Antibody Diluent with background-reducing components (Dako) for 1 h. Sections were incubated overnight with anti-VEGF, -angiostatin, and -endostatin antibodies at 4°C and then incubated for 1 h at room temperature with donkey anti-goat IgG HRP-bound secondary antibodies. DAB served as a chromogen, and Mayer’s hematoxylin was used for counterstaining.
RESULTS
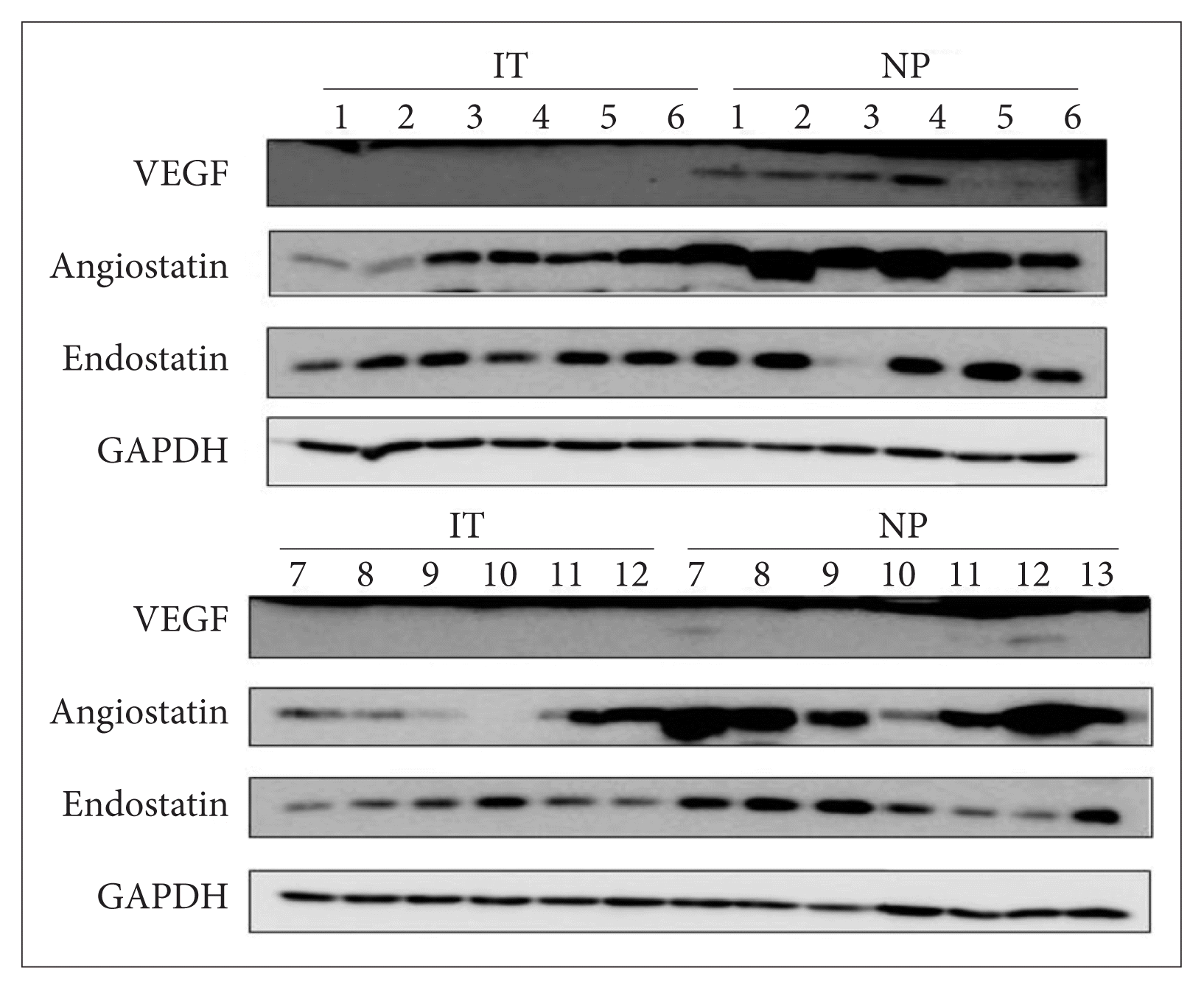
Levels of vascular endothelial growth factor, angiostatin, and endostatin protein
The relative VEGF protein level normalized to that of GAPDH was 3.07±2.79 in the NPs and 0.45±0.28 in the inferior turbinate mucosal samples. The figures for angiostatin were 3.81±2.34 and 0.93±0.83, while those for endostatin were 1.17±0.22 and 1.19±0.21, respectively (Fig. 1). Both the VEGF and angiostatin levels were higher in the NPs than in the control samples with statistical significance (p=0.001 for VEGF, p=0.001 for angiostatin), but the endostatin levels did not differ significantly (p>0.05) (Table 1).
The vascular endothelial growth factor/angiostatin and vascular endothelial growth factor/endostatin ratios
The VEGF/angiostatin ratio was 0.84±0.53 in the NPs and 2.21±5.34 in the control samples. However, the difference between two ratios was not significant (p>0.05). The VEGF/endostatin ratio was 2.81±2.57 in the NPs and 0.41±0.30 in the control samples, thus significantly higher in the NPs (p= 0.002) (Table 1).
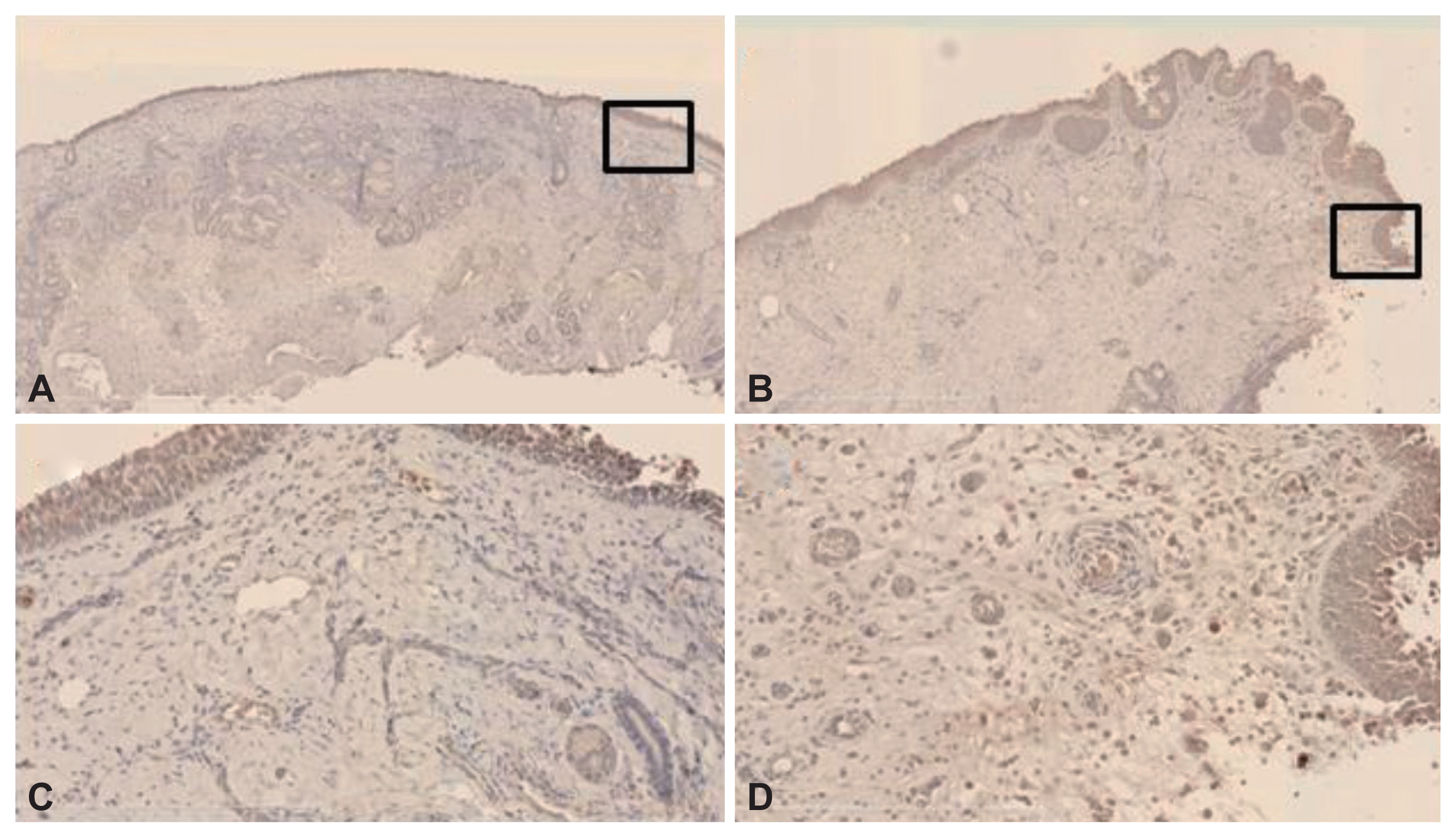
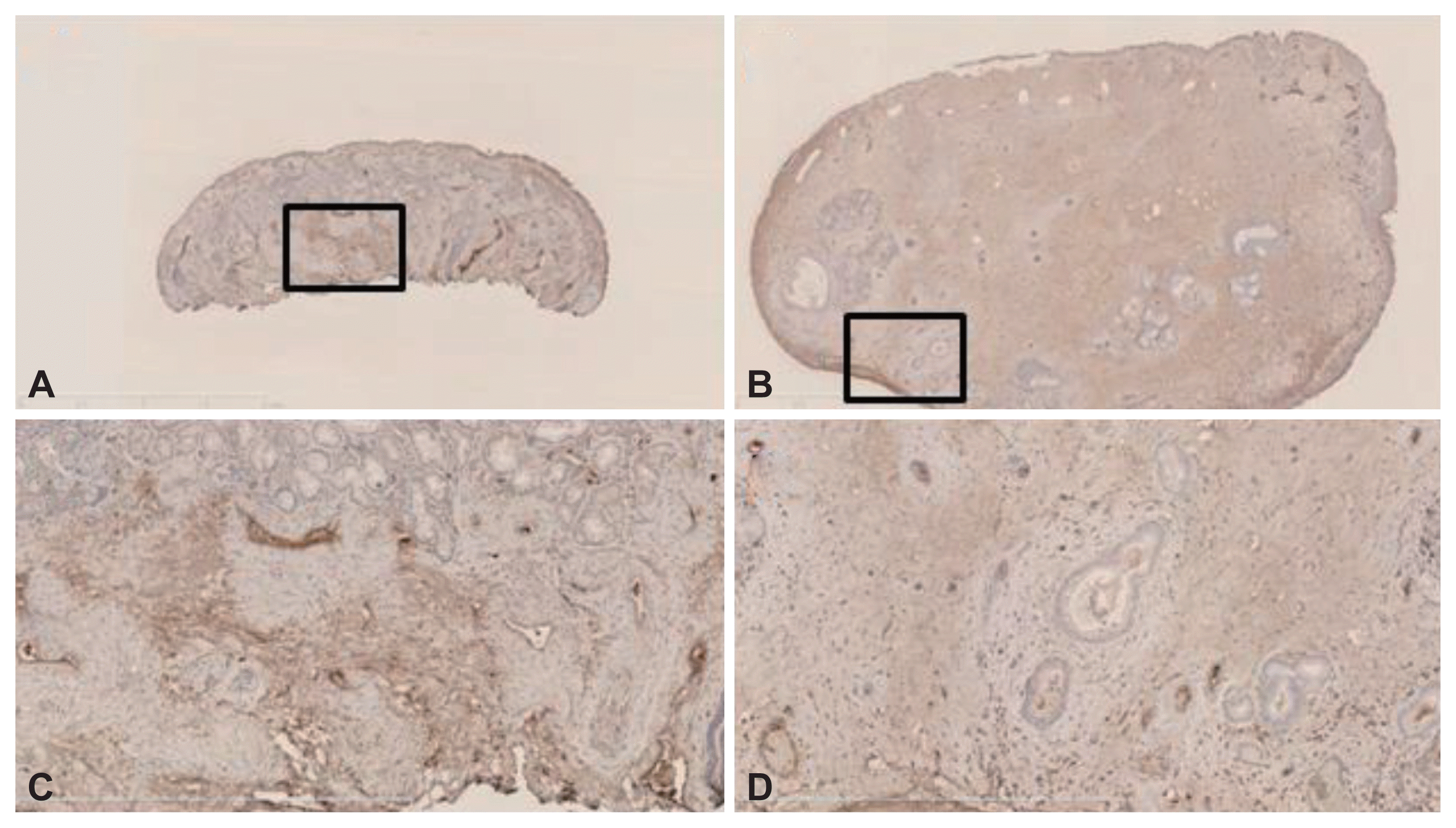
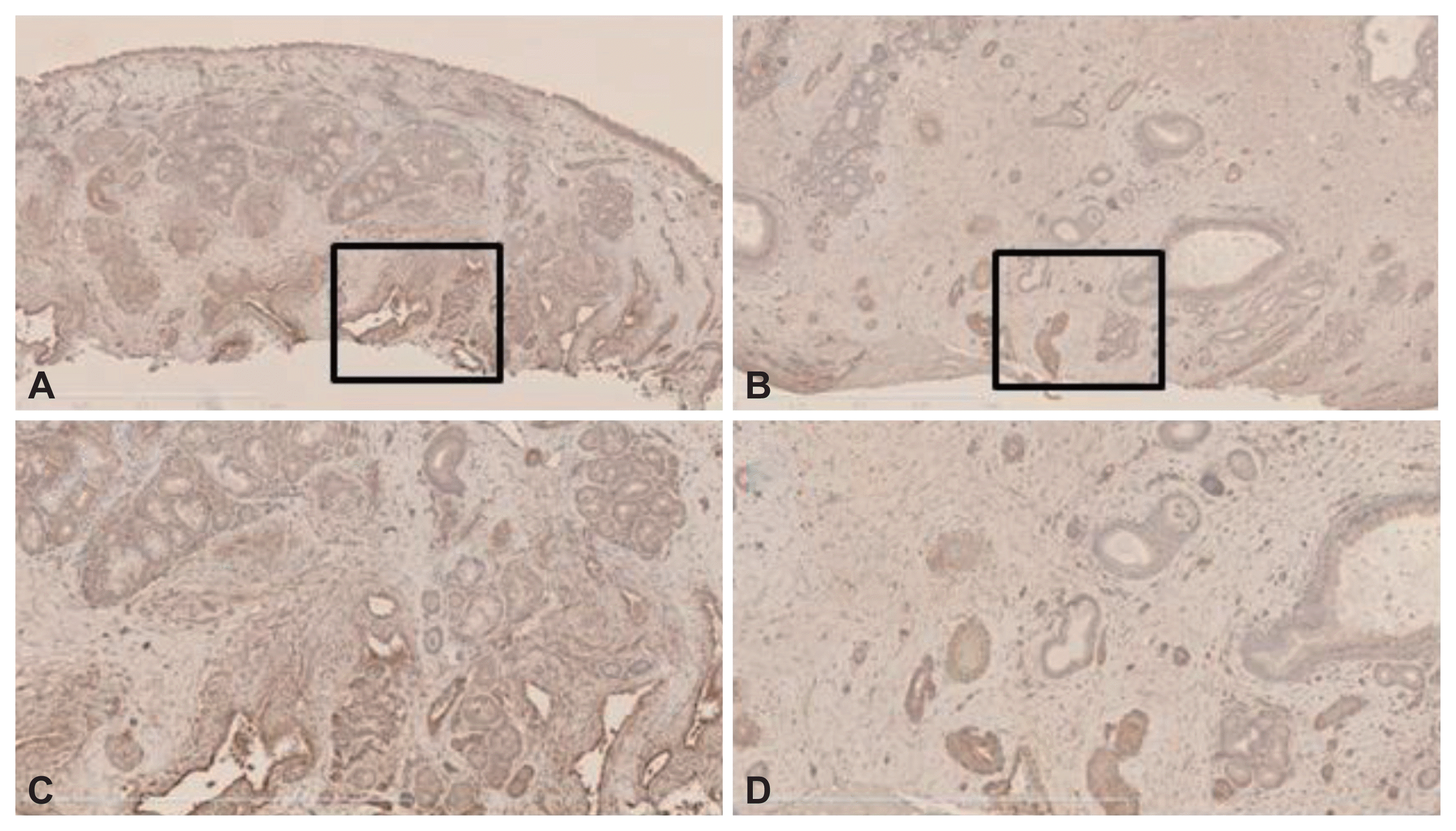
Immunohistochemical staining
VEGF immunoreactivity was evident principally in stromal cells, including ECs, fibroblasts, and inflammatory cells, in both the control and NP samples (Fig. 2). Angiostatin immunoreactivity was evident principally in the submucosal inflammatory cells of the stroma and ECs in both the control and NP samples (Fig. 3). Finally, endostatin immunoreactivity was strong in the ECs of stromal vessels in both samples (Fig. 4).
DISCUSSION
NPs are a chronic inflammatory disease in the mucosa of nose featuring infiltration by inflammatory cells (especially eosinophils) and tissue remodeling, including fibrosis, thickening of BM, and accumulation of extracellular matrix. Microvascular remodeling and angiogenesis are components of such remodeling in many chronic inflammatory diseases [17].
Of the many angiogenetic cytokines, VEGF is particularly important, being a major enhancer for vascular permeability in some inflammatory lesions [18,19]. VEGF could encourage microvascular permeability, contributing to the edema frequently evident in NPs. An increase in microvascular permeability triggers extravasation of plasma proteins participating in accumulation of the extracellular matrix, thereby accelerating NP growth [15]. We found that the VEGF level was higher in NPs than control samples.
Angiostatin induces EC apoptosis directly without any proliferative increase for compensation, resulting in a net reduction in EC numbers, in turn altering signaling by focal adhesion kinase and disrupting the normal turnover of such adhesions [20,21]. In addition, angiostatin decreases proliferation and migration of EC, and tube formation by inhibiting activation of VEGF- and the fibroblast growth factor-dependent extracellular signal-regulated protein kinases [22]. Angiostatin may exert its anti-angiogenic effects through binding to adenosine triphosphate (ATP) synthase in cell surface, thus depriving ECs of ATP [8]. However, in a mouse model of chronic hypoxia, angiostatin overexpression in lung tissue aggravated the development of pulmonary and right ventricular hypertension [23]. Also, hypoxia may play a role in NP initiation [24]. We found that the angiostatin level was higher in NPs than in control samples with statistical significance. We assume that chronic hypoxia in nasal cavity probably increase the angiostatin level, which may induce NP formation.
Endostatin, produced by various types of cells, is a strong endogenous inhibitor of angiogenesis [25]. Endostatin elevates apoptosis of EC by decreasing the expression levels of Bcl-xL and Bcl-2 (anti-apoptotic proteins) [12] and causing activation of caspase-3 [26]. Endostatin in extracelluar space inhibits the phosphorylation of signal-regulated protein kinases in fibroblast growth factor- and VEGF-induced EC cultures of artery, without hampering by the receptor binding of growth factors [12]. Also, endostatin reduces the production of a proinflammatory cytokine, tumor necrosis factor alpha [27]. Endostatin inhibits neoangiogenesis under various pathologies normally characterized by increased angiogenesis, including cancer [25], arthritis [27], and ulcerative colitis [28].
VEGF and endostatin frequently function as an inducer and inhibitor, respectively, of angiogenesis. The VEGF/endostatin ratio reflects the status of the angiogenic switch [29]. In sputum, the VEGF levels were higher in asthmatics than control subjects with significance. Moreover, the VEGF/endostatin ratio in asthmatics was higher than that in controls with significance [30]. Also, the endostatin level was significantly lower in seasonal allergic rhinitis patients exhibiting bronchial hyper-reactivity than in those lacking bronchial hyper-reactivity. We found that the VEGF level was higher in NPs than in controls with significance and the endostatin level did not differ between two groups. Also, the VEGF/endostatin ratio in NPs was significantly higher than in controls. We assume that a VEGF/endostatin imbalance in NPs means that hyper-reactivity in nasal mucosa can contribute to NP development.
Conclusion
The VEGF and angiostatin levels were higher in NPs than control samples. We suggest that nasal hypoxia elevates the angiostatin level, contributing to NP formation. Also, the elevated VEGF/endostatin ratio in NPs mean angiogenic switch due to hyper-reactivity, which may contribute to NP formation.
 XML Download
XML Download