

 PDF
PDF Citation
Citation Print
Print



INTRODUCTION
Fibrolamellar hepatocellular carcinoma (FLHCC) is a rare malignant hepatic cancer first reported in 1956 by Edmondson.1. Unlike classical hepatocellular carcinoma (HCC), FLHCC is common in young patients without any underlying liver disease and occurs in both women and men.2 This cancer type is rare in Asia, and only a few cases have been reported in Korea since the first case report in 1998.3-5 Since surgical resection is the only established effective treatment for FLHCC, early diagnosis and treatment are important. Here, we report a case of FLHCC in a young woman that was successfully treated by surgical resection. This case report was described according to the CARE guidelines available at https://www.care-statement.org/.
Go to : 
CASE REPORT
A 19-year-old woman without any underlying disease visited the outpatient clinic because of abnormal liver enzyme levels for the past 8 months. The patient had no chronic liver disease or viral hepatitis. She had no family history of HCC or other malignancies. She was not taking any medication, never smoked, and she drank a bottle of beer once a week. She had no abdominal pain, weight loss, or anorexia. The patient’s body mass index was 23, and there were no specific findings on physical examination. The results of the initial blood test were as follows: white blood cell count, 6,000/µL; hemoglobin, 13.8 g/dL; platelet, 438,000/µL; albumin, 4.2 g/dL; prothrombin time-international normalized ratio, 1.04; total bilirubin 0.6 mg/dL; alkaline phosphatase (ALP), 602 U/L; aspartate aminotransferase (AST), 66 U/L; and alanine aminotransferase (ALT), 106 U/L. The hepatitis B surface antigen, anti-hepatitis B surface antibody, and anti-hepatitis C virus antibody tests were all negative.
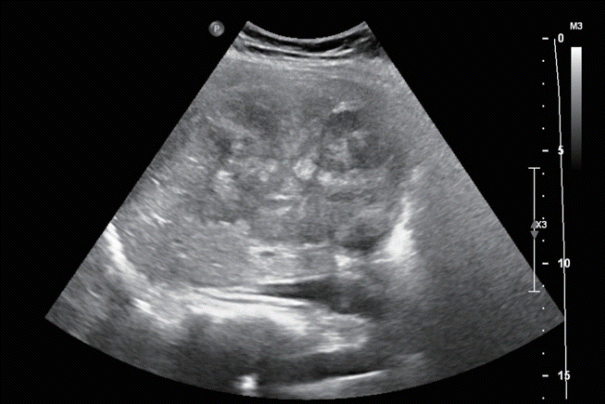
Abdominal ultrasonography showed an approximately 13 cm-sized lobulated mass with a suspicious central scar in the right liver (Fig. 1). Magnetic resonance imaging (MRI) revealed a lobulated liver mass with a central scar, prominent arterial enhancement, and low signal intensity in the delayed and hepatobiliary phases (Fig. 2). The levels of tumor markers were as follows: alpha-fetoprotein (AFP), 3.67 ng/mL; protein induced by the absence of vitamin K or antagonist-II (PIVKA-II), 316 mAU/mL; and carbohydrate antigen 19-9, 3.51 U/mL.
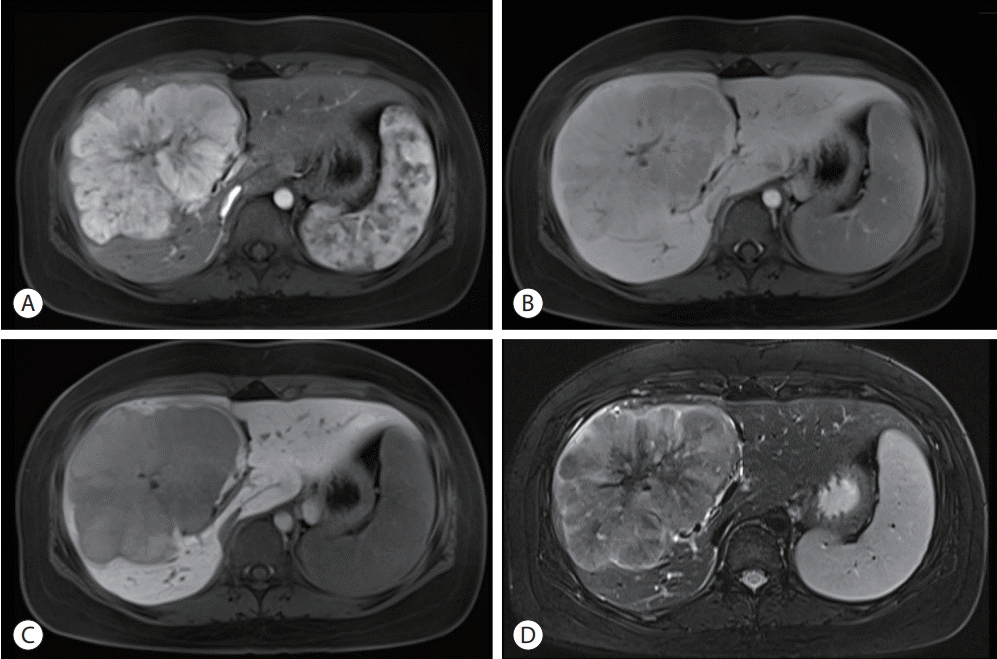 | Figure 2.Liver dynamic magnetic resonance imaging showed 13 cm sized lobulated iso-signal intensity mass in the right anterior section with prominent arterial enhancement (A) and low signal intensity in delayed phase (B), defect in hepatobiliary phase (C), and a central scar on T2 weighted image (D).
|
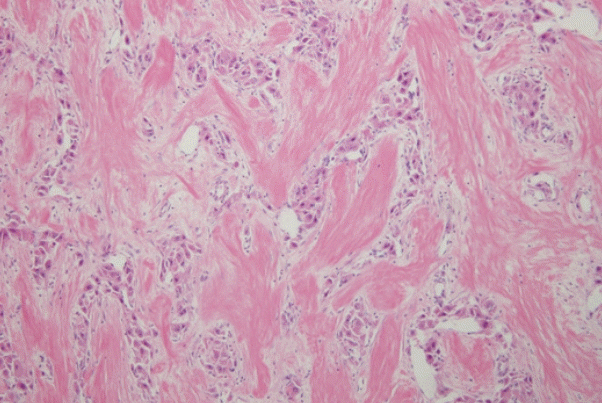
A liver biopsy was performed for accurate diagnosis. Pathology showed large polygonal cells with abundant eosinophilic cytoplasm and fibrous stroma arranged in parallel lamellae around the tumor cells (Fig. 3). The tumor was diagnosed as a FLHCC. There was no lymph node metastasis or distant metastasis on fluorodeoxyglucose-positron emission tomography scan. The tumor, node, and metastasis stage was Ib (T1bN0M0). An extended right hemihepatectomy was performed, and the surgical specimen also demonstrated the typical histological features of FLHCC, confirming its diagnosis. After surgery, AST, ALT, and ALP levels were normalized. The PIVKA-II level decreased to 28 mAU/mL and remained within the normal range. Follow-up computed tomography (CT) was performed every 3-6 months, and there was no recurrence for 3 years and 6 months.
Go to :
DISCUSSION
FLHCC is a rare liver tumor, accounting for <1% of all primary liver cancers and 13.4% of liver cancers occurring in individuals <40 years of age.6,7 This tumor is rare in Asia, with only a few cases being reported in Korea. FLHCC has several epidemiological characteristics that differ from those of classical HCC. It tends to occur in young people <40 years of age; it does not have a male predominance8; and clear risk factors have not been identified.7 Patients usually present with nonspecific symptoms, such as abdominal pain, abdominal distension, or tumors, and may be discovered incidentally without symptoms.9 In this case, the patient did not have any specific symptoms, and the tumor was discovered by performing abdominal ultrasonography due to elevated liver enzyme levels.
The diagnosis of FLHCC is based on clinical presentation and imaging studies. On CT, FLHCC often appears as a lobulated heterogeneously enhancing mass, and a central scar or calcification may be present. On MRI, tumors appear with hypointensity and hyperintensity on non-contrast T1- and T2-weighted images, respectively.10
AFP, an important tumor marker of HCC, is elevated in approximately 10% of FLHCC.9,11 Another tumor marker of HCC, PIVKA-II, is elevated more frequently. In a study that analyzed FLHCC in 41 patients, PIVKA-II was elevated in all 10 patients in whom that marker was measured.12 This case also showed similar findings: AFP was normal while PIVKA-II was elevated to 316 mAU/mL.
Pathology is the gold standard for the diagnosis. FLHCCs are characterized by large polygonal cells with abundant eosinophilic cytoplasm and abundant fibrous stroma arranged in parallel lamellae.2 The pathogenesis of FLHCC is unclear, although a unique fusion gene, DNAJB1-PRKACA (DNAJ/HSP40 homolog, subfamily B, member 1, and protein kinase, cAMP-dependent, catalytic, alpha), has been identified in nearly all FLHCC cases, and not in other HCC variants. A 400-kilobase deletion in chromosome 19 has been shown to result in a fusion gene, DNAJB1-PRKACA, which drives tumorigenesis.13 Until recently, the DNAJB1-PRKACA gene was considered to be specific for FLHCC. However, recent studies have reported that this fusion gene is also detected in other pancreatic and biliary tumors14, whereas FLHCC without the fusion gene has also been reported.15 We were not able to test this case for the DNAJB1-PRKACA alteration; however, the pathological features were typical of FLHCC and sufficient for its diagnosis. Further studies are needed to explain the roles of DNAJB1-PRKACA and other genes in the pathogenesis of FLHCC. Fibrolamellar-like features may be focally identified in otherwise conventional HCCs; such cases should not be diagnosed as FLHCC. Interestingly, HCCs with “focal fibrolamellar-like pattern” have been demonstrated to occur more commonly in older patients and to harbor BAP1 (breast cancer type 1 associated protein 1) gene abnormalities.16
Surgical resection is the primary treatment for FLHCC. Fiveyear survival rates following surgical resection range from 58% to 82%, and the recurrence rates range from 33% to 100%.17 Approximately 20% of the patients have unresectable tumors due to metastasis or major vessel involvement.9 These patients have poor outcomes, with 5-year survival rates from 0% to 5% and a median survival of <12 months.11,18-20 Systemic chemotherapy is administered to patients with unresectable tumors. Chemotherapeutic agents include cisplatin, fluorouracil, doxorubicin, gemcitabine, and irinotecan9; however, chemotherapy has not clearly demonstrated therapeutic efficacy. Studies on sorafenib treatment are limited; in one study, 10 out of 95 patients were treated with sorafenib, eight had disease progression after 2.5-7 months, one had a mixed response followed by progression, and one was lost to follow-up.9 The efficacy of other treatments, such as transarterial chemoembolization or external radiation therapy, has not been established.9
In this case, the persistently elevated liver enzyme levels of the patient led to an ultrasonographic evaluation, resulting in the detection of the FLHCC. Although the tumor was huge at initial diagnosis, it was successfully resected by surgery. Given the lack of effective alternative therapies, early diagnosis and surgical resection are important for the treatment of FLHCC.
Go to :
 XML Download
XML Download