

 PDF
PDF Citation
Citation Print
Print


서론
최근 환자 개인의 유전적, 환경적, 사회적 특성에 맞춰 진료하는 정밀의료가 점차 확대되고 있다. 당뇨병의 경우도 이러한 흐름에 맞추어 분류, 진단 및 치료에 대한 방침이 보다 구체화되어 가고 있다. 유전체 연구 및 DNA 염기 서열 검사법의 발전으로 최근 당뇨병의 유전적 원인 규명에 큰 발전이 있었다. 유전적 원인에 의한 당뇨병 중에서 하나의 DNA 염기서열변이에 의해 발생하는 단일유전자당뇨병(monogenic diabetes)의 경우 정밀의료의 주된 적용 분야가 될 것으로 기대되고 있다. 단일유전자당뇨병은 크게 MODY (maturi-ty-onset diabetes of the young), 신생아 당뇨병(neonatal diabetes mellitus), 증후군성 당뇨병(syndromic monogenic diabetes)으로 분류되고 있다. 그 중에서 MODY는 조기 발병, 상염색체 우성의 가족력, 비비만(nonobese) 체형, 발병 초기 인슐린 비의존성 등을 특징으로 하는 단일유전자당뇨병의 한 종류로 전체 당뇨병환자의 약 1˜3% 정도를 차지하는 것으로 알려져 있다[1].
MODY는 1970년대 처음 임상적으로 확인되었으며 이후 원인이 되는 유전자들이 규명됨에 따라 MODY 진단에 있어 유전자 검사의 역할이 확대되고 있다[2]. MODY는 그 원인이 되는 유전자에 따라 아형(subtype)이 구분되고 있으며 원인 유전자가 규명된 순서대로 OMIM (Online Mendelian In-heritance in Man) 및 여러 문헌에서는 MODY 1˜14로 구분하여 기술하고 있다. 하지만, 최근 Clinical Genome Re-source (https://clinicalgenome.org/affiliation/50016/) Monogenic Diabetes Expert Panel에서는 BLK, PAX4, KLF11 유전자는 단일유전자당뇨병을 일으킨다는 근거가 부족하다는 이유로 MODY 검사에서 해당 유전자를 제외해야 한다는 의견을 제시하고 있다[3,4]. 또한 MODY 아형 구분의 경우 기존에 사용하던 MODY 1˜14와 같은 숫자 분류 대신에 원인이 되는 유전변이에 따라 hepatocyte nuclear fac-tor-1 alpha (HNF1α)-MODY, glucokinase (GCK)-MO-DY 등과 같이 이름을 붙이고 그 종류를 구분하고 있는 추세이다.
본 글에서는 MODY의 분류 및 진단, 치료를 최근 진행된 연구 결과들을 바탕으로 정리해보고자 한다.
Go to : 
유병률
MODY와 관련된 연구들은 영국 Exeter 그룹을 중심으로 주로 서구권에서 이루어졌다. 독일, 영국, 미국 등에서 진행된 연구에 의하면 MODY의 발병률은 인구 100만 명당 24˜108명 정도로 추산되고 있으며 전체 당뇨 환자의 약 1˜3% 정도를 차지할 것으로 예측되고 있다[5–7]. 아시아에서는 상대적으로 MODY 에 대한 연구가 많이 이루어져 있지 않다. 최근 한국에서 1형당뇨병을 제외한 당뇨병환자 2,090명을 대상으로 진행한 연구에서 109명이 임상적으로 단일유전자당뇨병의 기준에 합당하였고 염기서열분석 결과 이 중 23명(21.1%)에서 병원성변이(pathogenic variant) 혹은 병원성가능변이(likely pathogenic variant)가 확인되었다. 최종적으로는 2,090명의 당뇨병환자 중 1.1%의 MODY 유병률을 보였고, 이는 외국에서 확인된 결과와 유사한 수준의 MODY 유병률이다[8].
MODY에 대한 연구가 가장 활발하게 진행되고 있는 영국에서는 인구 100만 명당 68˜108명 정도의 MODY 환자가 있을 것으로 추산하고 있다[9]. 이를 2020년 한국의 전체 인구수인 5,180만 명과 한국의 전체 당뇨병환자 약 494만 명을 대입해 계산해 보면 한국에는 약 3,522˜5,594명 정도의 MODY 환자가 있을 것으로 추산되고 이들 대부분은 제대로 분자유전학적 진단이 되지 않고 있는 것으로 생각된다.
Go to :
분류
단일유전자당뇨병은 그 발현 시기 및 동반되는 증상에 따라 MODY, 신생아 당뇨병, 증후군성 당뇨병으로 분류되고 있다. 그 중에서 MODY는 소아청소년기 당뇨병 발병, 상염색체 우성의 당뇨병 가족력, 비비만형 체형, 인슐린 비의존성 등을 특징으로 하는 유전적 질환이다. MODY는 그 빈도에 따라 비교적 흔하게 발생하는 MODY와 드물게 발생하는 MODY 로 나눌 수 있다(Table 1). 비교적 흔하게 발생하는 MODY로는 그 원인 유전자에 따라 GCK-, HNF1α-, HNF4α-, HNF1β-MODY가 있다. HNF1α, HNF4α는 hepatocyte nuclear factor 관련 유전자로 췌장 베타 세포의 발생과 분화 및 인슐린분비에 관여한다. 이러한 유전자의 변이에 의해 인슐린분비가 감소하여 당뇨병이 발생한다. GCK-MODY는 베타세포 포도당인산화효소(glucokinase)의 변이로 포도당에 의한 인슐린분비의 역치를 증가시켜서 공복혈당을 상승시키는 것으로 알려져 있다[10]. HNF1β-MODY는 신장 낭종 등 비뇨기계 이상을 동반할 수 있으며 전체 MODY의 약 1% 정도를 차지한다[11].
Table 1.
Maturity-onset diabetes of the young (MODY)-related genes and clinical characteristics [18]
Revised from the article of Yang et al. (Diabetes Metab J 2020;44:627-39) [18] under the Creative Commons Attribution Non-Com-mercial (CC BY-NC 4.0) license.
![]()
드물게 발생하는 MODY의 원인 유전자로는 IPF1 (PDX1), NEUROD1, CEL, INS, ABCC8, KCNJ11, APPL1 등이 알려져 있다. 이 중 ABCC8, KCNJ11은 MODY, 신생아 당뇨병, 신생아 고인슐린혈증에 의한 저혈당을 일으키는 유전자로 베타세포의 ATP민감포타슘통로(ATP-sensitive potassium channel)를 구성하는 단백을 표지한다. 만약 변이가 포타슘통로를 활성화시키면 당뇨병을 일으키고 억제시키면 고인슐린혈증이 발생한다[3].
신생아 당뇨병은 생후 6개월 이내에 진단되는 당뇨병으로 정의된다. 1형당뇨병과의 차이점은 1형당뇨병은 면역 기능이 활성화되기 이전인 생후 6개월 이내에는 잘 발생하지 않는다는 점이다. MODY의 유발 유전자로도 알려진 KCNJ11, ABCC8, INS의 변이가 신생아 당뇨병의 가장 주요한 변이로 확인되고 있으며 영구 신생아 당뇨병(permanent neonatal diabetes)의 약 50%를 차지하는 것으로 알려져 있다[12,13]. 그 외 일과성 신생아 당뇨병(transient neonatal diabetes)에서는 6q24의 과발현이 가장 흔히 확인되고 있다[13,14]. 다른 비교적 드문 형태의 신생아 당뇨병으로는 GATA6, PDX1, EIF2AK3, FOXP3를 포함한 유전자의 변이가 확인되고 있다.
증후군성 당뇨병(syndromic diabetes)은 유전변이와 관련하여 당뇨병뿐만 아니라 다른 여러 질환이 함께 발현되는 당뇨병을 의미한다. 대표적인 증후군성 당뇨병으로는 울프람증후군(Wolfram's syndrome)이 있다. 이는 WFS1 유전자의 변이에 의해 발현하는 질환으로 상염색체 열성 유전을 특징으로 하며 당뇨병, 청각장애, 시신경 위축, 요붕증 등을 동반하는 것으로 알려져 있다[15]. 그 외 미토콘드리아 유전자의 변이에 의해 발생하는 MIDD (maternally inherited diabetes and deafness)가 있다. 이는 주로 당뇨병, 청력저하, 신경학적 증상, 신기능 장애 등이 특징적으로 발생하는 증후군성 당뇨병으로 미토콘드리아 DNA 3,243 A> G의 변이에 의해 주로 발생한다.
Go to :
진단
최근 WES (whole exome sequencing), WGS (whole genome sequencing), targeted panel sequencing 등 유전자 검사법의 발전으로 MODY에서도 유전자 검사의 활용이 확대되고 있다. 하지만 당뇨병환자 중 어떤 환자에서 유전자 검사를 시행할지에 대한 선별기준에 대해서는 아직까지 정확하게 정립된 바 없다. 현재 활용되고 있는 MODY 환자의 임상적인 진단 기준으로는 ‘1) 당뇨병이 있는 가족 구성원 중 최소 1명은 진단 연령이 25세 미만임, 2) 상염색체 우성 유전 형태의 당뇨병이 3대에 걸쳐 확인됨, 3) 진단 후 최소 5년 동안 인슐린 치료를 받지 않았음, 4) 인슐린 수치가 정상범위임 (혈장인슐린 ≥ 2.0 µIU/mL or 혈장C-펩타이드 ≥ 0.6 ng/ mL), 5) 비만하지 않음(체질량지수 < 25 kg/m2)’의 다섯 가지 기준이 제시되고 있으며 유전자 검사를 위한 기준으로도 활용되고 있다[16].
영국에서 개발된 MODY calculator (https://www.diabetesgenes.org/exeter-diabetes-app/ModyCalculator)는 MODY가 의심되는 환자의 당뇨병 진단 연령, 성별, 체질량지수, 당뇨병 치료 방법, 인슐린 치료 시작까지 걸린 기간, 당화혈색소(HbA1c) 수치, 당뇨병 가족력 등 임상적 특징을 입력하면 환자가 MODY일 가능성을 계산해준다[17]. 이러한 MODY calculator를 유전자 검사가 필요한 MODY 환자를 선별할 때 활용해 볼 수 있다. 한국에서 진행된 연구에서 MODY의 유전변이가 확인된 경우 그렇지 않은 경우보다 MODY calculator를 이용하여 계산한 MODY 확률이 유의미하게 더 높다는 점을 보여주어 주로 백인을 대상으로 개발된 MODY calculator가 아시아인을 대상으로도 활용될 수 있음을 보여주고 있다[8].
Yang 등[18]은 당뇨병환자 중 MODY 유전자 검사가 필요한 경우에 대해서 다음과 같이 제시하고 있다(Fig. 1). 1형당뇨병의 경우 대개 생후 6개월 이후 발생하기 때문에 생후 6개월 이전 확인된 당뇨병의 경우 신생아 당뇨병에 대한 유전자 검사가 필요하다. 생후 6개월˜30세 이전에 1형당뇨병으로 진단된 환자 중 췌도자가항체가 없거나 C-펩타이드 수치가 비교적 높게(C-펩타이드 ≥ 6 ng/mL) 측정되는 경우에 MODY에 대한 유전자 검사를 진행할 것을 권고하고 있다. 그 외 30세 이전에 당뇨병이 발병한 환자 중 인슐린 저항성이 없으며 당뇨병의 가족력이 최소 2세대에 걸쳐 있고, 이 중 최소 1명 이상이 25세 이전에 당뇨에 진단된 경우 MODY 검사가 권고된다. 또한 특징적으로 비뇨기계의 기형이 있거나 신기능 악화가 동반되는 경우 HNF1β에 대한 유전자 검사가 추천된다. 당뇨병과 청력저하가 동반된 경우에는 MIDD나 울프람증후군과 같은 증후군성 당뇨병에 대한 검사를 해볼 것을 권고하고 있으며 임신당뇨병 환자의 경우 공복혈당이 다소 높아져 있으면서 당뇨병의 가족력이 있거나, 출산 후 2개월 이상 당뇨병이 지속되는 경우에 GCK-MODY에 대한 검사를 고려해보는 것이 권고된다.
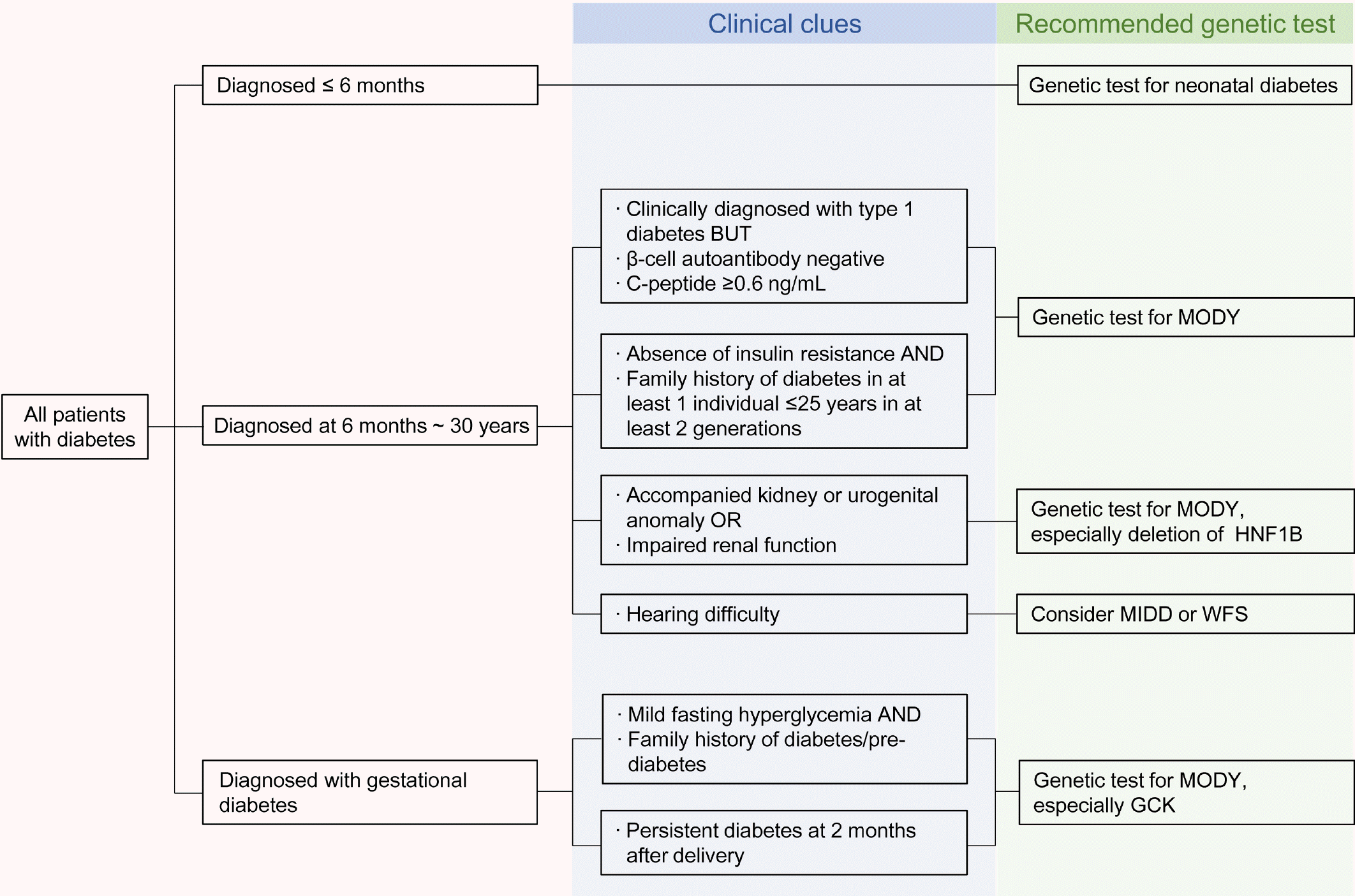 | Fig. 1.Approach to genetic testing based on clinical clues for monogenic diabetes. Adapted from the article of Yang et al. (Diabetes Metab J 2020;44:627-39) [18] under the Creative Commons Attribution Non-Commercial (CC BY-NC 4.0) license. MODY, maturity-onset diabetes of the young; HNFIB, hepatocyte nuclear factor 1B; MIDD, maternally inherited diabetes and deafness; WFS, Wolfram syndrome; GCK, glucokinase.
|
MODY를 확인하기 위한 유전자 검사가 늘어남에 따라 병원성이 없는 변이의 발견도 증가하고 있다. 따라서 발견된 변이가 단일유전자당뇨병의 원인이 되는 병인성변이인지 판정하는 것이 매우 중요하다. 발견된 변이가 병원성변이인지를 객관적으로 판정하기 위해 최근 ACMG-AMP (American College of Medical Genetics and Genomics and the Association for Molecular Pathology)에서 지침을 제시하고 있다[19]. 해당 지침에서는 각 변이를 병원성 증거(pathogenic evidence)와 양성 증거(benign evidence)의 조합에 따라 병원성(pathogenic), 병원성가능(likely pathogenic), 미확정(uncertain significance), 양성가능(likely benign), 양성(benign)으로 구분하여 질병 발병에 관여하는 정도를 보다 객관적으로 해석하도록 권고하고 있다[8,20,21]. 한국에서도 임상적으로 MODY 기준을 만족하는 환자 109명을 대상으로 targeted panel sequencing을 이용하여 원인 유전자를 확인하였고, 확인된 변이에 대해 ACMG-AMP 지침을 적용하여 제시함으로써 단일유전자당뇨병에서 ACMG-AMP 지침의 적용을 고려할 수 있음을 보여주었던 바가 있다[8].
Go to :
치료
MODY의 치료는 발병 원인이 되는 유전자 변이를 확인하고 이에 맞는 치료 방법을 선택하는 것이 핵심이다. 정확한 MODY의 진단과 치료는 환자뿐 아니라 환자의 가족에게까지 확대될 수 있다는 점에서 그 중요성이 강조된다. MODY는 비교적 젊은 나이에 발병하기 때문에 소아 혹은 청소년기에 1형당뇨병으로 잘못 진단되고 인슐린치료를 시작하여 장기간 유지되는 경우가 많다. 하지만 MODY의 경우 비교적 장기간 안정적으로 경구혈당강하제를 통해 조절할 수 있는 경우가 많다. 따라서 분자유전학적으로 MODY로 진단하면 이에 맞는 치료방법을 사용하여 환자의 삶의 질과 질병의 경과를 바꿀 수 있다.
HNF1α-, HNF4α-MODY의 경우 식후혈당이 주로 상승하는 것이 특징으로 알려져 있다[22]. HNF1α-, HNF4α-MO-DY는 설포닐유레아(sulfonylurea) 계열의 약제에 좋은 반응을 보여 이 계열의 약제를 1차적으로 사용하는 것이 권고된다. HNF1α-MODY를 대상으로 진행된 연구에서 설포닐유레아를 사용했을 때 1형당뇨병 대비 3.9배가량의 공복혈당 개선효과를 보인 바 있다[23]. 설포닐유레아의 부작용 중 하나인 저혈당을 줄이기 위해 HNF1α-MODY에서 GLP-1수용체작용제(glucagon-like peptide-1 receptor agonist)를 시도하는 연구들이 진행되었으며 설포닐유레아 대비 비슷한 혈당강하효과를 유지하면서 저혈당을 감소시키는 것으로 확인되어 설포닐유레아를 사용하기 어려운 환자에서 GLP-1수용체작용제를 고려해볼 수 있다[24]. 반면 HNF1β-MODY의 경우는 췌장의 형성부전과 인슐린저항성이 동반되어 있는 경우가 많아 초기 적극적인 인슐린 사용이 요구되는 경우가 많다.
GCK-MODY의 경우는 공복혈당이 상승하고, 식후혈당은 정상에 가깝게 유지되는 것이 특징이다. 고혈당 시 인슐린이 정상적으로 분비되는 경우가 많아 일반적인 경우에는 경구혈당강하제 등 특별한 치료 없이 경과 관찰이 가능하다. 다만 GCK-MODY 여성이 임신했을 경우에는 고혈당에 대한 치료가 필요할 수 있다. 실제로 임신당뇨병 환자의 약 1% 정도에서 GCK-MODY가 확인되고 있다[25]. 일반적인 임신당뇨병의 경우 모체의 고혈당이 태아의 인슐린분비를 촉진하여 태아의 과성장과 거대아(macrosomia)를 유발한다. 하지만 임산부와 태아 모두가 GCK-MODY일 경우에는 태아 또한 모체와 마찬가지로 포도당 농도에 따른 인슐린분비의 역치가 높아지기 때문에 태아에서 고인슐린혈증이 나타나지 않고 과성장 및 거대아의 빈도도 증가하지 않는다. 반면, 임산부는 GCK-MODY이나 태아는 정상인 경우에는 태아의 포도당 농도에 따른 인슐린분비 역치가 모체보다 낮아 과도한 인슐린분비와 과체중이 유발된다[26]. 따라서 GCK-MODY 임산부의 경우 태아의 유전형을 파악하는 것이 임신 중 고혈당의 치료 방향을 결정하는 데 중요한 역할을 한다.
최근에는 비침습적으로 모체의 혈장에 있는 세포 유리 DNA (cell free DNA)를 이용하여 태아의 유전자를 검사하는 기법이 개발되고 있다. 3명의 GCK-MODY 임산부를 대상으로 진행된 연구에서 모체의 세포 유리 DNA를 획득, 분석하여 태아의 GCK-MODY 유전변이 여부를 효과적으로 확인할 수 있음을 보여주었다[27]. 향후 세포 유리 DNA와 같은 검사 기법이 MODY 환자가 임신 시 비침습적으로 혈당 조절의 목표를 결정하는 새로운 대안이 될 것으로 기대된다.
Go to :
 XML Download
XML Download