

 PDF
PDF Citation
Citation Print
Print



INTRODUCTION
The goal of lipid-lowering therapy is to reduce the risk of atherosclerotic cardiovascular disease (ASCVD). Low-density lipoprotein (LDL) is a well-known causal factor of ASCVD [1]. Statins are administered as first-line agents to lower plasma LDL cholesterol (LDL-C) levels [2]. A number of outcome trials have demonstrated that statins have a consistent benefit in reducing the risk of ASCVD in primary and secondary prevention [3]. Therefore, current guidelines on the management of blood cholesterol recommend statin administration in all patients treated for secondary prevention, patients with familial hypercholesterolemia, patients aged 40 to 75 years with diabetes and plasma LDL-C ≥70 mg/dL, and patients treated for primary prevention without diabetes and with estimated 10-year ASCVD risk ≥7.5% [4,5]. However, despite optimal statin therapy, a significant residual ASCVD risk remains [6,7]. Therefore, there is a clinical need for novel agents which will help in lowering plasma LDL-C and other atherogenic particles effectively.
LDL-C LOWERING AGENTS
Several clinical and genetic studies have shown that LDL causes ASCVD, and ASCVD risk decreases in proportion to the degree and duration of LDL-C reduction [9–11].
PCSK9 inhibition by monoclonal antibodies
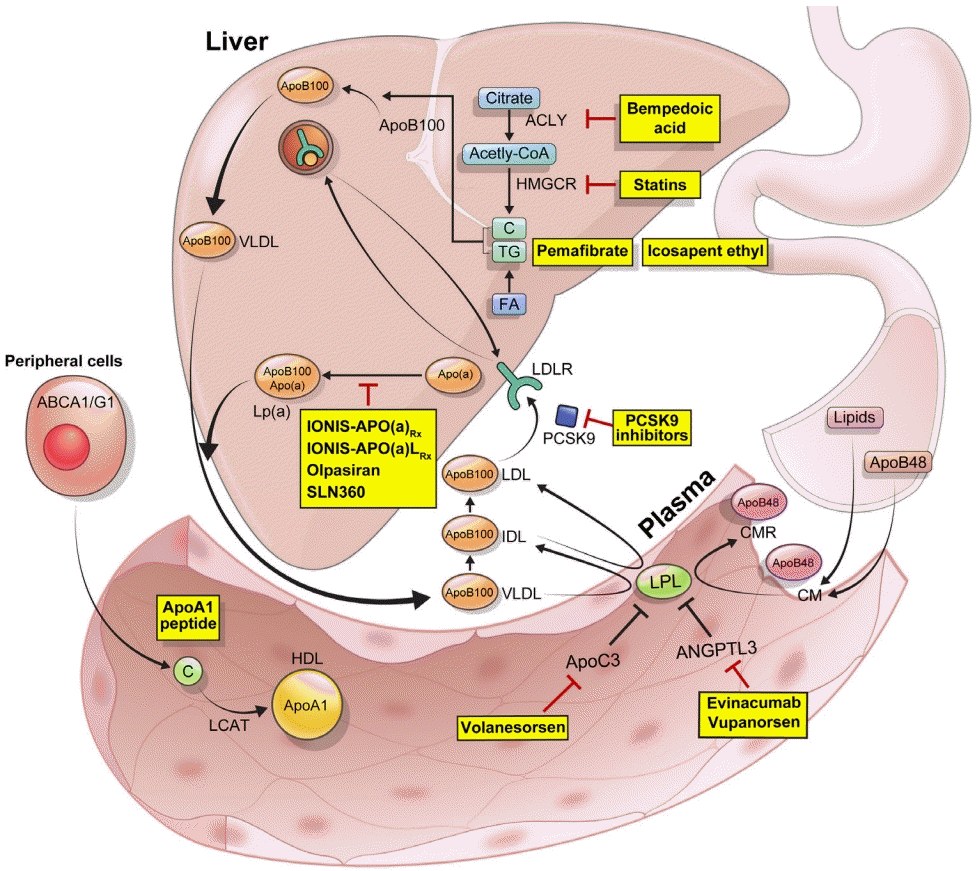
Proprotein convertase subtilisin/kexin type 9 (PCSK9), an enzyme predominantly produced in the liver, binds to the LDL receptor (LDLR) present on the surface of the hepatocytes, leading to its degradation and a subsequent increase in plasma LDL-C levels [12]. Thus, inhibition of PCSK9 causes an increase in LDLR number and a subsequent decrease in plasma LDL-C levels. Among the several monoclonal antibodies developed against PCSK9, evolocumab and alirocumab have been approved for clinical use and extensively evaluated in numerous clinical trials.
Evolocumab reduced plasma LDL-C levels by 53% to 75% whether administered as a monotherapy, used in conjunction with statin therapy, administered to patients with statin intolerance, or to patients with heterozygous familial hypercholesterolemia (HeFH) [13–17]. In patients with homozygous familial hypercholesterolemia (HoFH) who have defective LDLRs, evolocumab reduced plasma LDL-C levels by 31% [18]. In the Further Cardiovascular Outcomes Research With PCSK9 Inhibition in Subjects With Elevated Risk (FOURIER) trial involving patients with ASCVD, LDL-C ≥70 mg/dL or non-HDL-C ≥100 mg/dL while receiving statin therapy, evolocumab reduced the cardiovascular risk by 15% (hazard ratio [HR], 0.85; 95% confidence interval [CI], 0.79 to 0.92) [19].
Alirocumab reduced plasma LDL-C levels by 45% to 53% whether administered as a monotherapy, used in conjunction with statin therapy, or administered to patients with statin intolerance [20–23]. Depending on the genotype of the patient, alirocumab reduced plasma LDL-C levels by 39%–58% in patients with HeFH and by 11.9%–34.3% in patients with HoFH [24–26]. In the Evaluation of Cardiovascular Outcomes After an Acute Coronary Syndrome During Treatment With Alirocumab (ODYSSEY OUTCOMES) trial involving patients with an acute coronary syndrome, plasma LDL-C ≥70 mg/dL, non-HDL-C ≥100 mg/dL or apolipoprotein B (apoB) ≥80 mg/dL while receiving statin therapy, alirocumab reduced cardiovascular risk by 15% (HR, 0.85; 95% CI, 0.78 to 0.93) [27].
Current guidelines recommend the administration of PCSK9 inhibitors in patients with plasma LDL-C ≥70 mg/dL and high risk of ASCVD, while on maximally tolerated statin and ezetimibe therapy [4].
PCSK9 inhibition by RNA silencing
Inclisiran is a synthetic small interfering RNA (siRNA), which works by targeting the PCSK9 and is conjugated to triantennary N-acetylgalactosamine carbohydrates (GalNAc), which targets the siRNA to the liver [28]. In the Trial to Evaluate the Effect of Inclisiran Treatment on Low Density Lipoprotein Cholesterol (LDL-C) in Subjects With Heterozygous Familial Hypercholesterolemia (HeFH) (ORION)-9 trial involving patients with HeFH and plasma LDL-C ≥100 mg/dL while receiving statin therapy, a 300-mg dose of inclisiran sodium (corresponding to a 284-mg dose of inclisiran free acid) was administered on Days 1, 90, 270, and 450. On Day 510, inclisiran reduced plasma LDL-C levels by 47.9%. The inclisiran group reported a higher rate of injection-site reaction compared with the placebo group (17.0% vs. 1.7%). The majority of events were graded as mild and none were graded as serious [29]. In the United States-based ORION-10 trial involving a total of 1,561 patients with ASCVD, a 284-mg dose of inclisiran free acid was administered on Days 1, 90, 270, and 450. On Day 510, inclisiran reduced plasma LDL-C levels by 52.3%. The prespecified exploratory cardiovascular end-point occurred in 58 patients (7.4%) in the inclisiran group and 79 patients (10.2%) in the placebo group [30]. In the non-United States based ORION-11 trial involving a total of 1,617 patients with ASCVD or an equivalent, inclisiran reduced plasma LDLC levels by 49.9% on Day 510. The prespecified exploratory cardiovascular end-point occurred in 63 patients (7.8%) in the inclisiran group and 83 patients (10.3%) in the placebo group [30]. A meta-analysis of major cardiovascular events (MACE) studies involving PCSK9-inhibiting monoclonal antibodies or inclisiran showed that the results of the ORION 10–11 trials are in concordance with the results of 7 trials involving PCSK9-inhibiting monoclonal antibodies [31]. The ongoing phase-3 trial (ORION-4, NCT03705234) will help to further clarify the cardiovascular benefits of inclisiran.
Bempedoic acid
ATP-citrate lyase (ACLY) catalyzes the ATP-dependent conversion of citrate and coenzyme A (CoA) to oxaloacetate and acetyl-CoA. Acetyl-CoA, the precursor of 3-hydroxy-3-methylglutaryl-CoA (HMG-CoA), is crucial for the biosynthesis of cholesterol [32]. Thus, inhibition of ACLY leads to a reduction of acetyl-CoA and cholesterol synthesis, resulting in an increased number of LDLRs, causing a subsequent reduction of plasma cholesterol. Bempedoic acid is a small molecule that acts as a selective antagonist of ACLY. It is administered as a prodrug and requires activation by very-long-chain acyl-CoA synthetase-1, which is an enzyme mainly expressed in the liver. This property minimizes the exposure of the active drug to the non-hepatic tissue, such as the skeletal muscle [33].
In the Cholesterol Lowering via Bempedoic Acid, an ACL-Inhibiting Regimen (CLEAR) Harmony trial and the CLEAR Wisdom trial involving patients with ASCVD or HeFH, and plasma LDL-C ≥70 mg/dL while receiving statin therapy, bempedoic acid reduced plasma LDL-C levels by approximately 18% [34,35]. In the CLEAR Tranquility trial involving patients with statin intolerance and plasma LDL-C ≥100 mg/dL while receiving no more than low-dose statin therapy, bempedoic acid reduced plasma LDL-C levels by 28% [36]. In the CLEAR Serenity trial involving patients with statin intolerance and plasma LDL-C ≥130 mg/dL (for primary prevention) or plasma LDL-C ≥100 mg/dL (for secondary prevention and/or HeFH) while receiving various lipid-lowering therapies with selective cholesterol absorption inhibitors, bile acid sequestrants, fibrates, PCSK9 inhibitors, or niacin, either alone or in combination, bempedoic acid reduced plasma LDL-C levels by 21% [37]. A pooled analysis showed that bempedoic acid was associated with increase of blood urea nitrogen, creatinine, and uric acid. It also resulted in a decrease in hemoglobin. Gout incidence was higher in the bempedoic acid group compared with the placebo group (1.6/100 person-years [PY] vs. 0.5/100 PY). New-onset diabetes/hyperglycemia incidence was lower in the bempedoic acid group compared with that in the placebo group (4.7/100 PY vs. 6.4/100 PY) [38]. A meta-analysis showed that bempedoic acid significantly reduced plasma LDL-C levels by 14%, but no significant reduction was seen in MACE (17%) [39]. Interestingly, a Mendelian randomization analysis showed a reduction in cardiovascular disease (CVD) risk per unit decrease in the plasma LDL-C level in carriers of loss-of-function mutation in ACLY or HMGCR which mimicked the effect of ACLY inhibitors and statins, respectively [40]. A phase 3 cardiovascular outcome trial involving patients with statin intolerance (Evaluation of Major Cardiovascular Events in Patients With, or at High Risk for, Cardiovascular Disease Who Are Statin Intolerant Treated With Bempedoic Acid [ETC-1002] or Placebo [CLEAR Outcomes], NCT02993-406) is underway.
TG LOWERING AGENTS
Epidemiological, mechanistic, genetic, and clinical studies have consistently demonstrated that an elevated plasma TG level is associated with increased risk of ASCVD [41,42]. However, TG can be degraded by most cells in the body and, therefore, does not accumulate in the atherosclerotic plaque. Therefore, TG itself is unlikely the cause of atherosclerosis. Instead, TG-rich lipoproteins enter into the arterial intima and contribute to plaque formation, eventually leading to a high ASCVD risk [43].
American guidelines recommend the administration of fibrates or omega-3 fatty acids in patients with persistently elevated severe hypertriglyceridemia (TG ≥500 mg/dL) to prevent pancreatitis [4]. European guidelines recommend the administration of n-3 polyunsaturated fatty acids (icosapent ethyl 2×2 g/day) in combination with a statin in high-risk (or above) patients with TG levels between 135 and 499 mg/dL despite statin treatment [44]. The details of the icosapent ethyl trials will be discussed below.
Pemafibrate
Fibrates are well known TG-lowering drugs and are agonists of peroxisome proliferator-activated receptor alpha (PPARα). These include fenofibrate, bezafibrate, and gemfibrozil [45]. Previous randomized controlled trials (RCTs) have demonstrated inconsistent results on ASCVD risk reduction, particularly among patients on statins [46–50]. However, a meta-analysis demonstrated that fibrates reduce ASCVD risk in the subgroup of patients with high baseline plasma TG levels [51].
Pemafibrate is the first selective PPARα modulator. It shows more than 2,500 times stronger PPARα activation compared with fenofibric acid, the active form of fenofibrate. It also shows more than 5,000-fold selectivity for PPARα compared to PPARγ and more than 11,000-fold selectivity for PPARα compared to PPARδ [52].
In a phase 3 comparative trial involving Japanese patients with plasma TG levels between 150 and 500 mg/dL, pemafibrate and fenofibrate significantly reduced plasma TG levels by 46% and 39%, respectively. The pemafibrate group showed less frequent adverse events compared with the fenofibrate group (2.7% vs. 6.8%) [53]. In clinical trials involving Japanese patients with plasma TG ≥150 or 200 mg/dL while receiving statin therapy, pemafibrate reduced plasma TG levels by 50% from the baseline [54]. In a phase 2 trial involving a total of 408 statin-treated European patients with plasma TG levels between 175 and 500 mg/dL, the highest dose of pemafibrate tested, 0.2 mg twice a day, reduced plasma TG levels by 54.4% from the baseline [55]. A phase 3 cardiovascular outcome trial involving 10,000 patients with type 2 diabetes mellitus and plasma TG levels of 200 to 500 mg/dL while receiving statin therapy (Pemafibrate to Reduce Cardiovascular OutcoMes by Reducing Triglycerides IN patiENts With diabeTes [PROMINENT], NCT03071692) started from March, 2017 [56]. However, the study has been stopped early in April, 2022 for reasons of futility. Nevertheless, considering that pemafibrate may prevent disease progression in non-alcoholic fatty liver disease (NAFLD) patients with hypertriglyceridemia, the possibility that of ASCVD risk reduction by pemafibrate in patients with NAFLD remains [57]. Meanwhile, two clinical trials involving patients with plasma TG levels of 500 to 2,000 mg/dL (NCT03011450 and NCT03001817) are currently ongoing as well.
Omega-3 fatty acid preparations
Omega-3 fatty acids exert their effects through the PPAR-mediated pathway [58]. Even though omega-3 fatty acids have been recommended for patients with severe hypertriglyceridemia, most studies have not supported the benefits of omega-3 fatty acids in primary or secondary prevention of ASCVD in patients undergoing statin therapy [59–61]. Interestingly, in the Reduction of Cardiovascular Events with Icosapent Ethyl-Intervention Trial (REDUCE-IT) trial involving patients with plasma TG levels of 135 to 500 mg/dL, LDL-C levels of 41 to 100 mg/dL, and ASCVD or diabetes while receiving statin therapy, high dose (4 g/day) of icosapent ethyl, a form of eicosapentaenoic acid (EPA) reduced cardiovascular risk by 25% (HR, 0.75; 95% CI, 0.68 to 0.83) [62]. In the Effect of Vascepa on Improving Coronary Atherosclerosis in People With High Triglycerides Taking Statin Therapy (EVAPORATE) trial involving patients with coronary artery disease and plasma TG levels of 135 to 500 mg/dL while receiving statin therapy, 4 g/day of icosapent ethyl reduced low-attenuation plaque (LAP) volume by 17%, while in the placebo group LAP volume increased by 109% [63]. However, in the Outcomes Study to Assess STatin Residual Risk Reduction With EpaNova in HiGh CV Risk PatienTs With Hypertriglyceridemia (STRENGTH) trial involving patients with plasma TG levels of 180 to 500 mg/dL, LDL-C <100 mg/dL, and ASCVD or diabetes while receiving statin therapy, 4 g/day of omega-3 carboxylic acids containing EPA and docosahexaenoic acid (DHA) resulted in no significant reduction in the risk of cardiovascular events [64]. The contrasting results for icosapent ethyl versus omega-3 carboxylic acids have led to a controversy focused on design differences between the comparator oils (placebos) in REDUCE-IT trial and STRENGTH. REDUCE-IT used mineral oil whereas STRENGTH used corn oil. Importantly, the REDUCE-IT investigators reported significant increases in LDLC and CRP in the mineral oil group compared to icosapent ethyl-treated group [62]. That finding led to a cohort study, which was designed to mimic the REDUCE-IT trial; it showed that the contrasting CVD outcomes between the two trials could be partly explained by a difference in the effects of the comparator oils (mineral vs. corn), but not the active oils (EPA vs. EPA+DHA), on lipid traits and C-reactive protein [65]. Recently, a study of several ASCVD-related biomarkers in REDUCE-IT, conducted by the investigators themselves, showed that icosapent ethyl had minimal effects on those biomarkers, whereas levels increased among those allocated to mineral oil [66]. All together, these analyses suggest that the results of the REDUCE-IT trial must be interpreted with some caution.
ApoC3 inhibitor
Apolipoprotein C3 (apoC3) is a key regulator of TG metabolism. It is a potent inhibitor of lipoprotein lipase (LPL), the enzyme responsible for the lipolysis of TG in the very-low-density lipoprotein (VLDL) and chylomicron particles [67]. In addition, it inhibits hepatic clearance of VLDL and chylomicron remnants by LPL-independent pathways [68]. Plasma apoC3 levels are associated with an increased risk of CVD [69,70]. A previous study showed that participants heterozygous for loss-of-function mutation in the APOC3 gene showed a reduction of 49% in plasma apoC3 levels and of 35% in plasma TG levels along with very efficient in vivo lipolysis of VLDL TG [71]. A genome-wide association study (GWAS) showed that heterozygous carriers of a null mutation in the APOC3 gene had lower serum TG levels and reduced subclinical atherosclerosis than noncarriers [72]. In addition, loss-of-function mutations in the APOC3 gene are associated with 40% lower plasma TG levels and a 40% lower risk of CVD [73,74].
Volanesorsen (ISIS 304801; ISIS-APOCIIIRx) is a 2′-meth-oxyethyl–modified antisense oligonucleotide (ASO) targeting apoC3 mRNA [75]. IONIS-APOCIII-LRx (ISIS 678354; AKCEA-APOCIII-LRx) contains the same nucleotide sequence as volanesorsen, but contains an additional triantennary GalNAc complex that targets the ASO to the liver, allowing use of a much lower dose [76]. Volanesorsen has been tested in patients with elevated plasma TG levels and in patients with familial chylomicronemia syndrome (FCS), an autosomal recessive disease of chylomicron metabolism associated with severe hypertriglyceridemia and recurrent pancreatitis due to deficiencies of LPL, apolipoprotein C2 and A5, glycosylphosphatidylinositol-anchored HDL binding protein 1, and lipase maturation factor 1. In the A Study of Volanesorsen (Formerly IONIS-APOCIIIRx) in Patients With Familial Chylomicronemia Syndrome (APPROACH) trial involving patients with FCS having plasma TG ≥750 mg/dL, 300 mg/week of volanesorsen reduced plasma apoC3 levels by 84% and plasma TG levels by 77%. Seventy-seven percent of the patients in the volanesorsen group achieved plasma TG levels <750 mg/dL, compared to the 10% of the patients in the placebo group. Sixty percent and forty-five percent of the patients in the volanesorsen group exhibited injection-site reactions and thrombocytopenia with platelet levels of <100,000/μL, respectively [77]. In the A Study of Volanesorsen (Formerly ISIS-APOCIIIRx) in Patients With Hypertriglyceridemia (COMPASS) trial involving patients with plasma TG ≥500 mg/dL or FCS, 300 mg/week of volanesorsen reduced plasma TG levels by 71%, representing an absolute reduction of 869 mg/dL. In the volanesorsen group, 24% of the patients exhibited injection-site reactions, one patient exhibited thrombocytopenia with platelet level of <50,000/μL, and one patient exhibited serum sickness [78]. A meta-analysis of the available phase 2 and phase 3 clinical trials showed that volanesorsen significantly reduced plasma TG levels (68%), VLDL-C levels (73%), apoC3 levels (74%). It also raised plasma HDL-C levels by 40% and apoB levels by 8%. In 2019, volanesorsen was approved by the European Union (EU) for the treatment of adult patients with FCS [79]. A phase 2/3 trial of volanesorsen in patients with familial partial lipodystrophy is underway (NCT02527343).
In a phase 2 trial of patients with hypertriglyceridemia and an established ASCVD or high cardiovascular risk (NCT0338-5239), treatment with IONIS-APOCIII-LRx for 6 months resulted in plasma TG level reductions of 23% with a dose of 10 mg every 4 weeks, 56% with a dose of 15 mg every 2 weeks, 60% with a dose of 10 mg every 4 weeks, and 60% with a dose of 50 mg every 4 weeks, compared with an increase of 6% in the placebo group [80]. A phase 3 trial of IONIS-APOCIII-LRx in the patients with FCS is underway (NCT04568434).
ANGPTL3 inhibitor
Angiopoietin-like 3 (ANGPTL3), angiopoietin-like 4 (ANGPTL4), and angiopoietin-like 8 (ANGPTL8) inhibit LPL activity in a coordinated fashion, thereby regulating the plasma TG levels. ANGPTL3 and ANGPTL8 are produced and secreted by the liver, and ANGPTL8 activates ANGPTL3 to inhibit LPL activity in the heart and muscle [81,82]. ANGPTL4 is mainly expressed in the adipose tissues [83]. Under fasting conditions, ANGPTL4 expression is elevated and that of ANGPTL8 is suppressed, whereas expression of ANGPTL3 remains unaltered. Consequently, LPL activity is inhibited in the adipose tissues, but increased in the heart and muscle, thereby diverting the fatty acids and TGs away from the adipose tissues [84]. Under feeding conditions, ANGPTL4 expression is suppressed and that of ANGPTL8 is elevated, thereby leading to a restoration of LPL activity in the adipose tissues to take up TGs for storage [84,85].
ANGPTL3 regulates plasma TG and HDL-C levels by inhibiting LPL and endothelial lipase, respectively (Fig. 2). Loss-of-function mutations in the ANGPTL3 gene were associated with low levels of plasma TG, LDL-C, and HDL-C [86,87]. A previous study showed that participants heterozygous for ANGPTL3 loss-of-function mutation had 50% lower ANGPTL3 levels than noncarriers, and a 39% lower risk of coronary artery disease [88]. Another study showed that participants heterozygous for ANGPTL3 loss-of-function mutation had 17% lower plasma TG levels, 12% lower plasma LDL-C levels, and a 34% lower risk of coronary artery disease [89].
Evinacumab is a human monoclonal antibody that inhibits ANGPTL3. In the Evinacumab Lipid Studies in Patients with Homozygous Familial Hypercholesterolemia (ELIPSE HoFH) trial involving patients with HoFH and plasma LDL-C ≥70 mg/dL while receiving statin therapy, an intravenous infusion of evinacumab (15 mg/kg of body weight), every 4 weeks, reduced the plasma LDL-C levels by 49% and plasma TG levels by 50%. Even in patients with LDLR null-null variants, evinacumab reduced plasma LDL-C levels by 43.4%, as compared with the 16.2% increase in the placebo group. Adverse events were similar in both the groups, including the increase of liver fat on dose dependent manner [90]. In a phase 2 trial involving patients with refractory hypercholesterolemia despite on PCSK9 inhibitor and on maximal tolerated dose of statin, with or without ezetimibe (mean LDL-C of 150 mg/dL), evinacumab reduced plasma LDL-C levels by more than 50% at the maximum dose (450 mg/week administered subcutaneously and 15 mg/kg of body weight administered intravenously at intervals of 4 weeks) [91].
Vupanorsen (ISIS 703802; AKCEA-ANGPTL3-LRx; IONIS-ANGPTL3-LRx) is a GalNAc-conjugated ASO which targets ANGPTL3 mRNA. In a phase 1 trial, participants with plasma TG 90–150 mg/dL or >150 mg/dL were treated with a single dosage (20, 40, or 80 mg) or multiple dosages (10, 20, 40, or 60 mg per week for 6 weeks) of vupanorsen. After 6 weeks of treatment, participants in the multiple dosage group demonstrated a reduction in plasma ANGPTL3 levels (46.6%–84.5%), TG levels (33.2%–63.1%), LDL-C levels (1.3%–32.9%), VLDL-C levels (27.9%–60.0%), non-HDL-C levels (10.0%–36.6%), apoB levels (3.4%–25.7%), and apoC3 levels (18.9%–58.8%). There were no evidences of prothrombotic effects, bleeding episodes, significant reduction in platelet counts, or significant changes in the liver or renal function [92]. In a phase 2a trial, participants with plasma TG >150 mg/dL, type 2 diabetes mellitus, and hepatic steatosis were treated with 40 or 80 mg dosage of vupanorsen every 4 weeks, or 20 mg dosage every week. After 6 months of treatment, vupanorsen reduced plasma ANGPTL3 levels by 41% (when given dosage of 40 mg every 4 weeks), 59% (when given dosage of 80 mg every 4 weeks), and 56% (when given dosage of 20 mg every week); and plasma TG levels by 36% (when given dosage of 40 mg every 4 weeks), 53% (when given dosage of 80 mg every 4 weeks), and 47% (when given dosage of 20 mg every week). Vupanorsen, administered 80 mg every 4 weeks, reduced plasma apoC3 levels by 58%, remnant cholesterol levels by 38%, total cholesterol levels by 19%, non-HDL-C levels by 18%, HDL-C levels by 24%, and apoB levels by 9%. The most common adverse events reported were injection-site pruritus (in 14% of the participants) and injection-site erythema (in 12% of the participants). None of the participants exhibited a platelet level <100,000/mm3 [93]. In a phase 2b trial, participants with non-HDL-C >100 mg/dL and TG 150 to 500 mg/dL on statin therapy were treated with 80, 120, or 160 mg dosage of vupanorsen every 4 weeks, or 60, 80, 120, or 160 mg dosage every 2 weeks. After 6 months of treatment, vupanorsen reduced plasma non-HDL-C from 22.0% (when given dosage of 60 mg every 2 weeks) to 27.7% (when given dosage of 80 mg every 2 weeks). Injection site reactions and liver enzyme elevations were more common at higher doses. There was a dose-related increase in hepatic fat fraction [94].
ANGPTL4 and ANGPTL8 as targets
Studies of ANGPTL4 have shown controversial results. A previous study showed that the carriers of inactivating mutations in the ANGPTL4 gene had lower levels of plasma TG and a lower risk of coronary artery disease, compared with the noncarriers [95]. However, another study suggested that the circulating ANGPTL4 levels may not influence plasma TG levels or coronary heart disease risk [96]. Furthermore, a study showed an increased coronary heart disease risk among individuals having E40K mutation despite of having an athero-protective lipid profile [97]. Even though monoclonal antibodies against ANGPTL4 reduced atherosclerosis in mice and monkeys, the resulting severe inflammatory clinical phenotypes in these animals, such as peritonitis and mesenteric lymphadenopathy have precluded the use of ANGPTL4 antibodies in humans [95,98-100].
A few studies have suggested a potential benefit of ANGPTL8 inhibition. ANGPTL8-inhibiting monoclonal antibody significantly reduced the plasma TG levels and increased plasma HDL-C levels in mice and monkeys [101]. A genetic study showed that carriers of ANGPTL8 rs145464906T allele (stop-gain variant) had 15% lower plasma TG levels and 10 mg/dL higher plasma HDL-C levels compared with the non-carriers [102]. Another study showed that carriers of ANGPTL8 rs-760351239T allele (stop-gained variant) had 18.9 mg/dL lower plasma TG and 6.1 mg/dL higher plasma HDL-C levels in the UK Biobank, and had 24.0 mg/dL lower plasma TG levels and 9.1 mg/dL higher plasma HDL-C levels in the FinnGen Study [103]. In a phase 1 trial involving participants with mixed hyperlipidemia (TG ≥135 mg/dL and LDL-C ≥70 mg/dL), a single dose treatment of monoclonal antibody against ANGPTL3/8 complex resulted in dose-dependent decrease in TG, LDL-C, non-HDL-C, and apoB [104].
LIPOPROTEIN(A) LOWERING AGENTS
Lp(a) consists of an LDL-like moiety covalently linked to apolipoprotein(a) (apo(a)). The LDL-like moiety contains a central lipid core consisting of cholesteryl esters and TGs, an outer shell of phospholipids and unesterified free cholesterol, and an apolipoprotein B100 (apoB100) [105,106]. Apo(a) is highly homologous to plasminogen. Plasminogen consists of a tail domain, five kringle domains, and a trypsin-like protease domain, whereas apo(a) lacks the tail domain and the first three kringle domains present in plasminogen. Instead, apo(a) consists of 10 sequences homologous to plasminogen kringle 4 domain (K IV1–10), a kringle 5-like (K V), and a protease-like domain. Two key features of apo(a) is that K IV2 can be present in 1–40 copies, and the protease-like domain has no protease activity. Lp(a) also contains proinflammatory oxidized phospholipids (OxPLs) [107]. Therefore, Lp(a) can be atherothrombotic through several mechanisms: atherogenic via its LDL-like moiety, proinflammatory via its OxPLs content, and potentially antifibrinolytic via its apo(a) moiety that may bind to fibrin but has no fibrinolytic activity [108].
A meta-analysis and a prospective cohort study showed that the Lp(a) concentration was associated with coronary heart disease, stroke, and aortic valve stenosis [109,110]. A case-control study showed that two LPA variants were significantly associated with an increased plasma Lp(a) level and an increased risk of coronary disease [111]. In addition, the number of K IV2 repeats in apo(a), which was negatively correlated with the plasma levels of Lp(a), was also negatively correlated with the risk of myocardial infarction [112].
There is a lack of specific and potent therapies to lower Lp(a) levels in the plasma. The outcomes of lowering plasma Lp(a) levels pharmacologically in patients with ASCVD and high plasma Lp(a) levels are yet to be tested. The best evidence for potential benefits of lowering of plasma Lp(a) levels has come from studies in which patients have gone through lipid apheresis. In a prospective study involving patients with ASCVD and plasma Lp(a) >60 mg/dL while receiving lipid-lowering therapy, apheresis lowered the incidence rate of cardiovascular events by 70%–80% [113]. However, there was no control group, and LDL-C levels were also reduced. In another study involving patients with coronary artery disease and Lp(a) >60 mg/dL while receiving lipid-lowering therapy, apheresis lowered the incidence rate of cardiovascular events by 80%–90% [114]. Because subgroup of patients with LDL-C >100 or ≤100 mg/dL had similar reductions in rate of major adverse coronary events, the effect of LDL-C on outcome could be excluded [114].
Apo(a) inhibitor
So far, no therapeutic agent has been approved for specifically lowering Lp(a). However, an agent targeting apo(a) has been developed. IONIS-APO(a)Rx (ISIS-APO[a]Rx) is a 2′-O-methoxyethyl-modified ASO targeting apo(a) [115]. IONIS-APO(a)LRx (Pelacarsen; AKCEA-APO[a]LRx; TQJ230) is a modified IONIS-APO(a)Rx which is conjugated with a triantennary GalNAc complex, and, because it is efficiently targeted to the liver, shows more than 30-times higher potency than the parent ASO [116].
In a phase 1 trial involving healthy adults with plasma Lp(a) ≥10 mg/dL, IONIS-APO(a)Rx resulted in significant lowering of plasma Lp(a) levels in a dose-dependent manner: 39% in the 100 mg group, 59% in the 200 mg group, and 77% in the 300 mg group [115]. In a phase 2 trial involving participants with plasma Lp(a) levels between 50 and 175 mg/dL (cohort A) or those with Lp(a) ≥175 mg/dL (cohort B), IONIS-APO(a)Rx resulted in plasma Lp(a) level reduction of 67% and 72%, respectively [116].
In a phase 1/2a trial involving participants with plasma Lp(a) ≥30 mg/dL, IONIS-APO(a)LRx resulted in Lp(a) level reduction of up to 92% [116]. In a phase 2 trial involving patients with ASCVD and Lp(a) >60 mg/dL, IONIS-APO(a)LRx was administered in ascending doses at intervals of 1 to 4 weeks. After 6 months of treatment, plasma Lp(a) levels were reduced by 35% at a dose of 20 mg every 4 weeks, 56% at 40 mg every 4 weeks, 58% at 20 mg every 2 weeks, 72% at 60 mg every 4 weeks, and 80% at 20 mg every week, as compared with the 6% with the placebo. There were no significant differences between both the groups in terms of the adverse events that occurred, such as fluctuation in platelet counts, liver and renal toxicity, and influenza-like symptoms. Notably, 27% of patients in the IONIS-APO(a)LRx group exhibited injection-site reactions [117].
A phase 3 cardiovascular outcome trial of IONIS-APO(a)LRx in the patients with ASCVD event and plasma Lp(a) >70 mg/dL (Lp(a)HORIZON, NCT04023552) is underway.
Recently, the results of olpasiran, an siRNA to target LPA were reported. In a phase 1 trial involving participants with Lp(a) 70–199 nM or >200 nM, single doses of olpasiran at 3, 9, 30, 75, or 225 mg were administered. Plasma Lp(a) levels were reduced in a dose-responsive manner from 71% to 97%. Only one patient on olpasiran experienced an injection site reaction [118].
In addition, the results of SLN360, an siRNA to target LPA messenger RNA were reported. In a phase 1 trial involving participants with Lp(a) >60 mg/dL and no known CVD, single doses of SLN360 at 30, 100, 300, or 600 mg were administered. Plasma Lp(a) levels were reduced by 46% at a dose of 30 mg, 86% at 100 mg, 96% at 300 mg, and 98% at 600 mg, as compared with the 10% with the placebo. Low-grade injection site reactions and headache were common treatment-emergent adverse events [119].
HDL TARGETING AGENTS
HDLs are heterogenous subpopulations of discrete particles that differ in density, size, shape, and composition [120]. Plasma HDL-C level is an excellent predictor of ASCVD risk [121]. However, a Mendelian randomization study failed to show a correlation between plasma HDL-C levels and ASCVD risk [122]. Indeed, few epidemiological studies showed increased mortality when plasma HDL-C was elevated [123,124]. In addition, RCTs with niacin and cholesteryl ester transfer protein inhibitors, in which HDL-C levels were significantly increased, failed to show a reduction in ASCVD risk [125,126]. Thus, the importance of HDL function, rather than HDL-C level, has been suggested [121].
ApoA1 peptide
Apolipoprotein A1 (apoA1) is a protein synthesized in the liver and intestine and functions as the major structural component of HDL [127]. Lipid-free apoA1 triggers microsolubilization of cell membrane lipids, facilitating transfer of free cholesterol and phospholipids, after an interaction with the ATP-binding cassette transporter A1, to form nascent HDL particles [128,129]. These nascent HDL particles, after being remodeled by the lecithin cholesterol acyltransferase, transform into mature HDL particles. These mature HDL particles interact with the ATP-binding cassette transporter G1, ATP-binding cassette transporter G4, and scavenger receptor class B type 1 to mediate additional cholesterol efflux from the foam cells in the arterial wall [130,131].
Researchers have considered HDL mimetics containing apoA1 as a potential treatment to exploit the athero-protective effects of HDL [132]. However, RCTs of two different reconstituted apoA1 products showed disappointing results. In the MDCO-216 Infusions Leading to Changes in Atherosclerosis: a Novel Therapy in Development to Improve Cardiovascular Outcomes-Proof of Concept IVUS, Lipids, and Other Surrogate Biomarkers (MILANO-PILOT) trial involving patients with an acute coronary syndrome, 20 mg/kg of body weight of a recombinant variant apoA1 (A1-Milano; MDCO-216) weekly showed no regression of plaque volume compared to that by the placebo [133]. In the CER-001 Atherosclerosis Regression Acute Coronary Syndrome Trial (CARAT) trial involving patients with an acute coronary syndrome, 3 mg/kg of body weight of a recombinant wild-type apoA1 (CER-001) weekly showed no regression of plaque volume compared to that by the placebo [134]. A phase 3 trial of CSL-112, a novel formulation of native apoA1 purified from the human plasma, in the patients with acute coronary syndrome is underway (NCT0-3473223) [135].
CONCLUSIONS
The goal of a lipid-lowering therapy is to reduce the risk of ASCVD. Statins are first-line agents to lower plasma LDL-C, a well-known risk factor of ASCVD. However, even under optimal statin therapy, a significant residual ASCVD risk remains. Therefore, novel drugs other than statins and novel targets other than LDL-C are definitely necessary to reduce the risk of ASCVD.
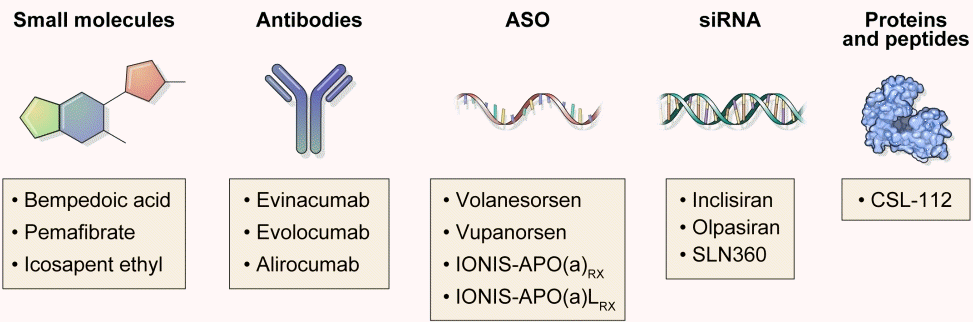
Emerging methods from human genetics, such as GWAS, Mendelian randomization study, and novel platforms for drug discovery such as RNA-targeted therapies have contributed significantly to the development of diverse classes of lipid-lowering agents (Fig. 2). Inclisiran, the siRNA which targets PCSK9, shows comparable effects to that of PCSK9 monoclonal antibodies. Bempedoic acid, an ACLY inhibitor, lowers plasma LDL-C levels and is a valuable treatment option for the patients with statin intolerance. Pemafibrate, the first selective PPARα modulator, shows a favorable benefit-risk balance compared to fenofibrate in early phase trials but seems to have failed to reduce ASCVD in PROMINENT. Based on the REDUCE-IT trial, high dose icosapent ethyl shows promise as a viable treatment option. Evinacumab, the ANGPTL3-inhibiting monoclonal antibody, reduces plasma LDL-C levels in the patients with refractory hypercholesterolemia who had been treated with maximum tolerated doses of statins and other lipid-lowering therapies such as PCSK9 inhibitors. ASOs that target apoC3, ANGPTL3, and Lp(a) have significantly attenuated dyslipidemic states. ApoA1 mimetic peptide is considered as a potential treatment to exploit the athero-protective effects of HDL-C, but needs more supporting evidences. We hope that these novel lipid-lowering agents can be used in real clinical settings in the near future.
 XML Download
XML Download