

 PDF
PDF Citation
Citation Print
Print



INTRODUCTION
The pituitary gland, often called the “master gland” orchestrates multiple effector hormonal organs and other glands by secreting various tropic hormones, namely growth hormone (GH), follicle stimulating hormone (FSH), luteinizing hormone (LH), thyroid stimulating hormone (TSH), adrenocorticotrophic hormone (ACTH), prolactin, oxytocin (OXT), and vasopressin. It is well-known that effector hormones, particularly thyroid hormone, cortisol, insulin-like growth factor 1 (IGF1), and sex hormone (i.e., estrogen and testosterone) play a significant role in skeletal modeling and remodeling. We have learned over the past decade that pituitary hormones also directly exert skeletal actions. The reciprocal relationship with pituitary and effector hormones poses a significant challenge in differentiating direct from indirect actions of pituitary hormone, but sophisticated genetic modification and small molecule intervention without altering systemic levels of effector hormone levels has allowed us to examine the direct skeletal role of these glycoproteins. These non-traditional skeletal effects of pituitary hormones will be discussed in this review.
Go to : 
UBIQUITOUS EXPRESSION OF PITUITARY HORMONE RECEPTORS
The hypothalamic-pituitary-endocrine (i.e., adrenal, thyroid, and ovaries/testis) feedback axis is truly fascinating physiology that establishes the central concept of endocrinology. The physiology of endocrine system and pathological changes from the imbalance—hormonal excess or insufficiency and their skeletal complication are well studied.
Over the past years, it has become increasingly clear that the pituitary tropic hormones also exert non-traditional actions bypassing the endocrine axis. Many studies have now documented the ubiquitous expression of the G-protein coupled receptors (GPCRs) for pituitary hormones, establishing that these receptors are not solely localized to endocrine organs. For example, in mammals, the TSH receptors (TSHRs) are expressed in the pituitary gland, thymus, testis, kidney, brain, adipose tissue, bone and heart albeit at lower levels [1,2]. The genetically modified mice with replacement of exon 1 of the TSHR with green fluorescent protein (GFP) visualized non-thyroidal expression of TSHR [3,4]. TSHR expression has been documented previously in osteoblast-like rat osteosarcoma cells (UMR106 cells), rodent calvaria-derived primary osteoblasts, pre-osteoblastic MC3T3-E1 cells, human osteoblast-like cells, osteoclast precursors, and mature osteoclasts [2,5-9].
Likewise, apart from their expression in granulosa cells of the ovary and Sertoli cells of the testis [10-12], FSH receptors (FSHRs) are present on bone, fat and the nervous system [4,13-15]. In the human female reproductive tract and placenta, FSHR is expressed in vascular endothelial cells, endometrial glands, cervical glands and stroma, stromal cells and muscle fiber, and interstitial macrophage from testis [16,17]. Furthermore, in endothelial cells of various cancer tissues (i.e., breast, colon, pancreas, kidney etc.), FSHR expression was much higher than gonadotropin cells [18]. Also, chondrocyte-like ATDC5 cells expressed functional FSHRs [19]. Interestingly, our data using RNAscope showed FSHR expressions in cerebral cortex and hippocampus, particularly in granular layer and pyramidal tract [13]—this suggested a role for FSH-FSHR interactions in cognition. An FSHR type 2 isoform without exon 9 is found in human monocytes and osteoclasts at a lower levels compared with ovarian cells. Of note, FSHR has 10 exons with the first 9 exons encoding extracellular domain and exon 10 expressing transmembrane domain [20-22].
ACTH receptor (MC2R), one of five melanocortin receptors (MC1R–MC5R), is expressed predominantly in the adrenal glands and required for adrenal gland development and steroidogenesis [23], is also expressed in bone cells—osteoblast-like cells (MG63), SaOS2 cells, and normal human osteoblasts (NHOS). ACTH and other proopiomelanocortin (POMC)-derived peptides increased cyclic adenosine monophosphate (cAMP) levels in NHOS [24]. Interestingly, immune cells, namely macrophages, splenic B and T-cytotoxic (CTL) cells were capable of secreting ACTH [25], and osteoclasts, which are differentiated from hematopoietic stem cells, not only expressed and processed POMC and secreted ACTH, but also expressed MC2R [24], suggesting possible autocrine or paracrine actions of ACTH in bone remodeling.
Other pituitary hormones also have their receptors expressed in bone and bone cells. The findings of prolactin receptors on osteoblasts in cell culture and bone tissue suggested possible direct effect of prolactin in bone remodeling independently of concomitant hypogonadism in the setting of hyperprolactinemia [26-28]. OXT and vasopressin, which are secreted from posterior pituitary gland, also have their receptors expressed in osteoblasts and osteoclasts [29]. The functional relevance of the pituitary hormone receptor expression in skeletal tissue and bone cells will be reviewed in the following section.
Go to :
HIGH FSH AND BONE LOSS
FSH was first isolated from sheep pituitary gland and FSH injection increased the size of ovarian follicles in hypophysectomized rats suggesting its reproductive role [30]. Likewise, after receiving partially purified FSH preparation, amenorrheic women showed increased urinary estrogen levels and increased the size of the uterine cavity and ovaries [31]. Subsequently, it was shown that FSH-containing cells are present in the pituitary gland. A negative feedback loop between FSH and estradiol was subsequently documented [32-35].
Our group first described an effect of FSH in skeletal remodeling using genetically modified FSHR and FSHβ mice. Haploinsufficient mice for both the ligand and receptor had normal estrogen levels and developed an intact uterus—yet they showed higher bone mass compared with wild-type mice, essentially separating the action of FSH from that of estrogen [36]. Knock-out mice were also protected, but in Fshr−/− mice, this protection due to absent FSHR signaling was confounded by the associated hyperandrogenemia [37]. A recent study performed µCT analysis of distal femurs from male and female Fshb+/+, Fshb+/−, and Fshb−/− mice at 8 months of age and showed higher bone volume and less trabecular spacing in the absence of Fshβ [38].
Using interventional approaches, lowering FSH in the ovariectomized rat by gonadotropin-releasing hormone (GnRH) agonist (leuprolide) decreased the number of osteoclasts and bone loss area in alveolar bone [39]. Likewise, blocking the FSH effect using anti-FSHβ antibodies decreased osteoclast differentiation up to approximately 20% [40]. More importantly, blocking with anti-FSHβ antibody attenuated ovariectomy-induced bone loss [4,14,15,40]. Furthermore, we found that the skeletal effect of FSH was independent of other hormones, including estrogen, testosterone, inhibin, or activin. This finding was consistent with a recent study where humanized monoclonal anti-FSHβ antibody (MS-Hu6) treatment reduced osteoclast formation, and did not alter LH, GnRH, or testosterone levels [41]. Complementing these data, it was shown that an FSH-glutathione-S-transferase (GST) fusion protein prevented ovariectomy-induced bone loss in rats [42].
FSH acts on the bone FSHR isoform that is coupled to the G-protein Gi2α, and in doing so, increases osteoclastogenesis and bone resorption and suppresses bone formation [20,36,40,43]. FSH also enhances RANK expression and indirectly promotes osteoclastogenesis by stimulating the release (or enhancing the receptor expression) of tumor necrosis factor α (TNFα), interleukin (IL)-1β, and IL-6 [20,21,44-46]. FSH-induced osteoclast differentiation is also abolished in bone marrow macrophages from mice lacking immunoreceptor tyrosine-based activation motif (ITAM) adapters (Fig. 1) [47].
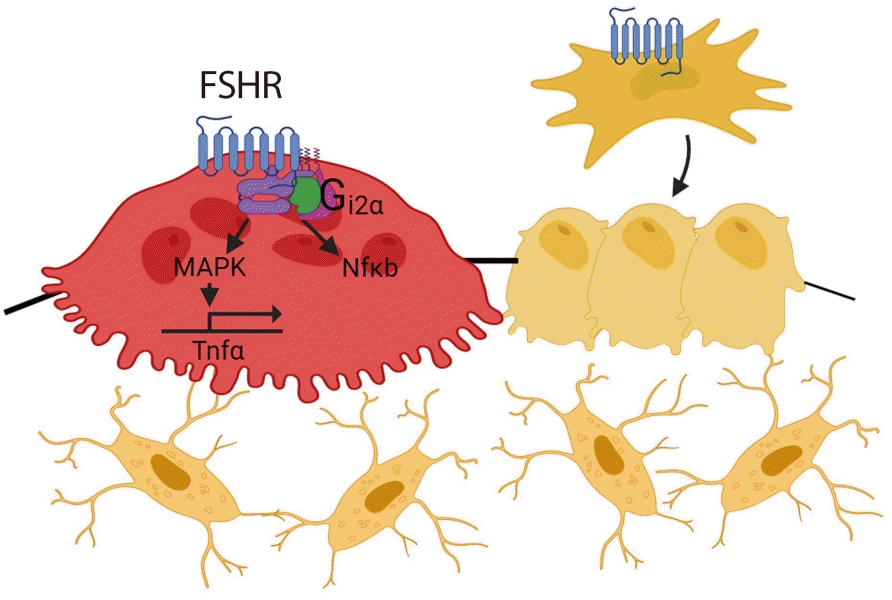 | Fig. 1.Follicle stimulating hormone (FSH) receptors are expressed in osteoclasts and mesenchymal stem cells. FSH/FSH receptor (FSHR) isoform binding activates nuclear factor κB (NFκB) and mitogen-activated protein kinase (MAPK), upregulates tumor necrosis factor α (Tnfα), and results in increased osteoclastogenesis and decreased osteoblast differentiation.
|
FSH negatively regulates osteoblastic bone formation. FSHRs are expressed on mesenchymal stem cells (not mature osteoblasts), and FSH antibody treatment yields greater osteoblast precursor colony counts similar to mesenchymal cells isolated from Fshr null mice [20,43]. MS-Hu6 antibody treatment also showed increased bone formation with increased mineralization apposition rate (MAR) and up-regulated osteogenic genes (Alpl, Col1a1, and Runx2) [41].
Supporting the mouse studies, multiple observational studies have examined associations between bone parameters and serum FSH. Notably, bone mineral density (BMD) of lumbar spine (LS), femur, and forearm show a negative correlation with an increase in serum FSH level, although these findings were limited due to the reciprocal relationship of FSH and E2 (High FSH and low E2) [48,49]. Other studies have attempted to examine the association after controlling for the estrogen effect. A study that sub-categorized the peri- and post-menopausal healthy women with irregular menstruation by FSH levels showed that participants with high FSH (>40 mIU/mL) had lower BMD compared with subjects with lower FSH levels (<40 mIU/mL) [50]. FSH increase by 1 standard deviation (SD) was associated with a 5% decrement in LS BMD in pre-, peri-, and post-menopausal women when E2 levels were similar in pre- and peri-menopausal women, but 2-fold higher FSH levels in the latter [51]. Bone turnover markers (BTMs) (i.e., urinary deoxypyridinoline [D-Pyr], urine N-terminal telopeptide [NTx], urinary C-terminal telopeptide [CTX], serum osteocalcin, and bone alkaline phosphatase) were shown to correlate with FSH positively, suggesting high bone turnover with elevated FSH levels [51-53].
Comparing skeletal changes in patients with hypothalamic functional amenorrhea versus hypergonadotropic amenorrhea provided another line of evidence for a direct FSH effect. In patients with premature ovarian failure who were younger than the age of 40, those with hypergonadotropic amenorrhea (FSH >40 IU/L) had lower LS BMD than patients with hypogonadotropic amenorrhea despite having other risk factors for low BMD, such as nutritional deficit and underweight [54].
The most relevant clinical correlations between elevated FSH and bone loss were noted in the Study of Women’s Health Across the Nations (SWAN), a longitudinal cohort of 2,375 perimenopausal women with diverse ethnic backgrounds. There was a strong positive correlation with serum FSH and NTx and osteocalcin independently of estrogen levels, and FSH was a stronger predictive marker for rapid bone turnover during perimenopausal period than estrogen [55]. Other large cohorts from China and Iceland (AGES-Reykjavik Study of Older Adults) also noted an inverse correlation between FSH and BMD [55-57]. In addition, the National Health and Nutrition Examination Survey (NHANES) III cohort showed a strong negative correlation between serum FSH and femoral neck (FN) BMD in perimenopausal women. In post-menopausal women, an incremental increase in FSH was associated with almost three-fold higher risk for osteoporosis, although the investigators used body mass index as a surrogate marker for estrogen status [58]. The bone turnover range of normality (BONTURNO) study similarly showed that women with high FSH levels (>30 IU/L) had high BTMs despite having regular menstruation [59]. Moreover, women with the activating FSHR polymorphism (rs6166) displayed lower bone mass and high resorption markers [60]. All of findings above suggest that the FSH surge during perimenopausal period might itself drive high bone turnover and bone loss. The selective inhibition of FSH action using antibodies such as ours may allow a further delineation of the effects of high FSH versus low estradiol.
Go to :
TSH AND BONE: NOT JUST THYROXINE
Multiple observational studies have documented an association between low TSH levels and a low bone mass. Again, the reciprocal relationship between TSH and thyroid hormone (T4/T3) poses a challenge in understanding the independent skeletal effect of TSH. Most studies therefore used healthy euthyroid or subclinical hyperthyroid subjects.
Analysis of the Rotterdam study showed a positive correlation between serum TSH levels and FN BMD among euthyroid subjects [61]. NHANES (1999 to 2002) showed similar results where low-normal TSH levels (0.39 to 1.79 mIU/L) were associated with a higher risk of osteoporosis independently of thyroid hormone levels [62]. Studies from China and Korea also noted similar patterns of associations [63-65].
Associations of TSH levels with fracture risk is more challenging as fracture itself is a multi-factorial process with many confounding factors. The Osteoporosis and Ultrasound Study (OPUS) study showed a lower risk of fracture with higher TSH levels among euthyroid subjects [66]. However, other large epidemiology studies, namely the Tromsø study and the Rotterdam study did not find a clear correlation between TSH levels and fracture risk [61,67]. Lastly, a meta-analysis including thirteen studies of euthyroid adults showed an almost 1.25 times higher risk of hip fracture in a group with lower TSH levels (0.45 to 0.99 mIU/L) compared with subjects with high-normal TSH (3.50 to 4.49 mIU/L) [68].
The negative skeletal effect of low TSH becomes more prominent when examining subjects with subclinical hyperthyroidism. The Study of Osteoporotic Fracture (SOF) showed that increased risk of fracture with a lower titer of TSH; participants with serum TSH (≤0.1 mIU/L) had a three-fold higher risk of hip fracture compared with normal TSH levels (0.5 to 5.5 mIU/L) [69]. The Odense Patient data Explorative Network Thyroid Status and Register Outcomes (OPENTHYRO) registry cohort also showed that low TSH levels (<0.3 mIU/L) was associated with a higher risk of hip fracture with 45% increase per 1 SD decrease of TSH [70]. Two separate meta-analyses summarizing 13 and six studies, respectively, again showed the hip fracture risk was significantly higher and BMD was lower in subclinical hyperthyroidism [71,72]. In addition, several studies suggested that micro-skeletal structure was suboptimal in elderly patients with subclinical hyperthyroidism and in patients under TSH suppression treatment following thyroidectomy for thyroid cancer [73-75]. However, this correlation of TSH and bone mass was not as robust in subclinical hypothyroidism, pre-menopausal women or men as in post-menopausal women [76].
Several studies have also documented changes in BTMs after recombinant human TSH (rhTSH) injection. CTX and NTX levels dropped within days and procollagen type 1 N-terminal propeptide (P1NP), a marker of bone formation increased without changes in thyroid homorne levels [77-79].
These human correlations point to a direct effect of TSH on bone, which was first described by us using TSHR haploinsufficient (Tshr+/–) mice. They displayed significant bone loss, despite intact thyroid follicles and normal thyroid hormone levels [2]. Homozygotic Tshr–/– mice were runted and had decreased BMD, which persisted despite thyroid hormone replacement from the birth [2,80]. The key characteristics of skeletal changes were summarized as increased bone turnover with predominantly increased osteoclast-driven bone resorption with focal sclerosis reminiscent of Paget’s bone disease [2]. In a later study, iatrogenic hyperthyroidism was induced by T4 pellet implantation after pre-treatment with carbimazole. Greater bone turnover and bone loss followed in Tshr–/– hyperthyroid mice compared with hyperthyroid wild-type, suggesting that the absence of TSH signaling contributed further bone loss in the setting of hyperthyroidism [81]. The skeletal phenotype of other genetically modified mice Tshrhyt/hyt with a loss-of-function mutation in TSHR (TSHRP556L) and Pax8–/–mouse, which lacks a transcription factor for thyroid follicle development were summarized in the recent reviews [76,82].
The skeletal effect of TSH arises from a direct action on osteoclast TSHRs, but also indirectly through TNFα [80,83], an osteoclastogenic cytokine that is known to cause bone loss [45]. Immune-mediated TSH action on skeleton was demonstrated by interventional studies using TNFα antibody and genetic modification. Mice with TSHR deficiency showed increased osteoclastogenesis with up-regulated TNFα expression, but without any change in receptor activator of NF-κB-ligand (RANKL) or macrophage-colony-stimulating factor (M-CSF), two important signals for osteoclastogenesis [2]. Moreover, treatment with an anti-TNFα neutralizing antibody or crossing Tshr–/– mice with Tnfa–/– mice reversed increased osteoclast differentiation [2,80,83]. These findings are relevant to clinical observations, where patients with hyperthyroidism are accompanied by elevated TNFα and soluble TNF receptor (TNFR) levels [84]. Mechanistically, TSH inhibits high-mobility group box proteins (HMGB) 1 and 2 expression and their binding to DNA, and suppresses JNK1/2 and IĸBα phosphorylation and c-jun and p65 nuclear translocation, all of which are critical steps in TNFα synthesis [2,85]. Likewise, TSHR overexpression decreases the activator protein (AP)-1 and nuclear factor κB (NFκB) binding in response to RANKL and TNFα/IL-1 [83].
An immune-skeletal-endocrine interaction of TSH is further supported by the identification of a splice variant of TSH (TSH-βv) in bone marrow cells [86,87]. Bone marrow-derived murine and human macrophages, and CD11b+ and other immune cells were shown to express TSH-βv [87,88]. This finding, in part, explains how TSHR in skeletal tissue can be functional despite being expressed at low levels [7,8].
Interestingly, the regulation of TSH-βv does not follow traditional hypothalamic-pituitary-thyroid axis feedback. TSH-βv injection into mice increases serum T4/T3 levels, but TSH-βv expression in leukocytes was not increased or suppressed in response to thyrotropin releasing hormone (TRH) or T3, respectively [89]. Also, T4/T3 regulated TSH-βv expression positively at mRNA and protein levels rather than suppressing it [87,90]. In addition, pro-inflammatory cytokines suppressed pituitary TSH secretion [91], but at the same time, trafficked immune cells expressing TSH-βv to the thyroid glands [92]. These findings indicate that immune cells such as macrophage in bone marrow might have the potential to attenuate the negative skeletal effects of low TSH and high T4/T3 in pro-inflammatory hyperthyroidism by secreting TSH-βv. This proposed TSH-related circuitry involving the bone-immune-endocrine axis needs further study (Fig. 2).
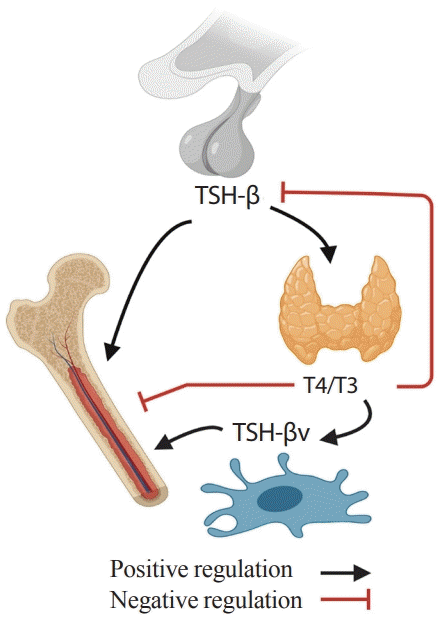
Unlike the anti-osteoclastic effect, TSH effects on osteoblasts are not as straightforward. In bone marrow-derived primary osteoblast cultures, TSH suppressed colony forming units and osteoblast gene expression, which was associated with downregulated vascular endothelial growth factor (VEGF) receptor FLK-1 and the Wnt co-receptor LRP5 [2]. However, other studies using calvaria-derived osteoblast, UMR106 and SaOS2 cell lines, show that TSH can stimulate osteoblastogenesis [6,93] through the activation of protein kinase Cδ and upregulation of the noncanonical Wnt components frizzled and Wnt5a [94]. In addition, co-culture of macrophages and osteoblasts enhanced osteoblastogenesis, which was attenuated in the presence of anti-TSH-βv antibody—this reaffirmed local skeletal effect of TSH-βv [87]. In vivo, intermittent low dose rhTSH injections induced bone formation and bone gain and recovered bone loss after ovariectomy [9]. Whether the anabolic action of TSH is dependent on the dose or the frequency similarly to parathyroid hormone needs to be further studied.
Furthermore, TSHR might interact with IGF1 receptor (IGF1R). TSH up-regulated IGF1 and IGF2 expression as well as stimulatory IGF-binding proteins [93]. TSHR and IGF1R synergistically increase osteopontin through crosstalk between the receptors in a signaling complex through β-arrestin 1, scaffolds linking G-protein-coupled receptors to extracellular signal-regulated kinases 1/2 (Erk1/2) signaling [95]. TSH induces β-arrestin-1 binding to TSHRs, β-arrestin then phosphorylates Akt1, p38, and Erk1/2 and upregulates Alp, Rankl, and Opn. In contrast, downregulation of β-arrestin 1 inhibits TSH-mediated osteogenic gene upregulation [93,95,96]. In all, it seems clear at least experimentally—with observational evidence from large epidemiologic cohorts—that the net bone loss in hyperthyroidism is the result of interplay between components of the TSH-skeletal-immune axis, mediated by selective actions of T4/T3, TSH, and TSH-βv.
Go to :
ADRENOCORTICOTROPIC HORMONE AND BONE
ACTH directly effects bone bypassing glucocorticoid action. Our group showed that ACTH treatment in rabbit with methylprednisolone-induced avascular osteonecrosis of femoral head (AVFN) reduced necrotic surface, which likely was mediated by upregulating VEGF expression [97]. VEGF upregulation by ACTH was also shown in a later study [98]. In addition, ACTH enhances protease inhibitor alpha-2-macroglobulin (A2M), which likely promotes osteoblastic differentiation through transforming growth factor β induction (Fig. 3) [99]. These findings suggest that ACTH can be a therapeutic target for treating AVFN, which is often caused by vascular insufficiency from trauma, alcohol and more importantly, long-term steroid use [99,100]. However, patients with ACTH-dependent Cushing disease had less bone loss compared with those with ACTH-independent adrenal Cushing’s syndrome [101].
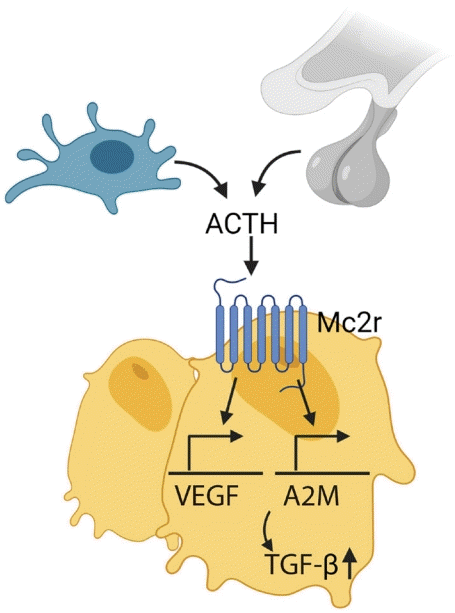 | Fig. 3.Adrenocorticotropic hormone (ACTH), secreted by pituitary gland and immune cells (i.e., macrophage), acts on melanocortin 2 receptor (Mc2r) on osteoblasts and upregulates vascular endothelial growth factor (VEGF) and alpha-2-macroglobulin (A2M), which induces transforming growth factor β (TGF-β).
|
A later study using Mc2r−/− mice showed increased cortical bone thickness without a change in trabecular bone. Serum osteocalcin levels were high in the homozygotes, while urinary deoxypyridinoline (D-Pyr) was decreased. However, unfortunately, the global knock-outs were confounded with adrenal insufficiency with deficiencies in glucocorticoid, mineralocorticoid, and catecholamines [102]. The skeletal phenotype of bone cell specific MC2R deficient mice has not been reported yet.
Go to :
THE MAIN PLAYER: GH OR IGF1
While GH directly acts on bone through a GPCR, its action predominantly occurs through IGF1 [103]. IGF1 is synthesized mainly in the liver and approximately 80% circulates bound to IGF-binding protein-3 (IGFBP3) and the acid labile subunit (ALS). Studies using GH, GH receptor (GHR) and IGF1 deficient mice provided evidence of independent and common effects of GH versus IGF1 on the skeleton. The double Ghr/Igf1 knock-out mouse showed more severe growth retardation compared with either GHR deficient or IGF1 deficient mice [104]. Studies using liver-specific IGF1 and bone-specific IGF1 mouse delineated the systemic and local effects of IGF1. Unlike complete IGF1 knock-out mice, liver-specific IGF1-deficient mice, despite significant drop in circulating IGF1 (~75%), developed and grew normally, except displaying suboptimal cortical bone quality [105,106]. However, further lowering systemic IGF1 level below ~10% in liver-specific IGF deficient mice by deleting IGFBP3 and the ALS resulted in marked growth retardation [107,108]—this suggests that certain levels of systemic IGF1 are required. On the other hand, bone-specific IGF1 deficient mice with normal circulating IGF1 level showed significant growth retardation, low bone mass, impaired bone formation and mineralization [109]. Together, GH, systemic IGF1, and local IGF1 play concerted roles in postnatal skeletal growth, modeling and remodeling.
Go to :
PROLACTIN AND BONE MASS
Prolactin receptor deficient (Prlr–/–) mice, generated by deleting exon 5 of the Prlr gene showed lower BMD in both sexes. Dynamic histomorphometry showed decreased bone formation. Of note, estrogen levels in female Prlr–/– mice were significantly lower compared with wild-type littermates, but testosterone in males did not differ. These findings can thus partly be explained by hypogonadism, but interestingly osteoclast surfaces, a hallmark of hypogonadal bone loss, were not different in Prlr–/– mice compared using wild-type littermates [28]. In contrast, a gain-of-function study in which hyperprolactinemia was induced by anterior pituitary transplantation in the setting of ovariectomy showed that prolactin stimulated bone turnover with net bone resorption. It was thought to be through decreased osteoprotegerin (OPG) expression in osteoblasts, which increased RANKL/RANK and osteoclastogenesis [27].
Given its physiological role in lactation and procreation, the effect of maternal prolactin in embryonic skeletal development was examined. A study measured alkaline phosphatase as an indirect marker for bone turnover after prolactin administration during pregnancy. Newborn pups from treated dams showed almost ~30% decrease in alkaline phosphatase and reduced bone formation, without any change in calcium or parathyroid hormone levels [26]. This in vivo finding was consistent with in vitro osteoblast cell cultures using primary osteoblast and MG63 cells, where Alp and Ocn were downregulated and alkaline phosphatase activity was suppressed after prolactin treatment [26,27]. Whether prolactin exerts a protective bone effect by downregulating bone turnover in the setting of excessive maternal bone resorption needs to be further studied [110,111].
Go to :
OXYTOCIN AND INTERGENERATIONAL CALCIUM TRANSFER
OXT is a nonapeptide that is synthesized in hypothalamus and released via the posterior pituitary gland. OXT mediates milk ejection and uterine contraction during parturition, and regulates social behavior centrally [112,113]—but we have shown that it plays a role in calcium homeostasis and bone remodeling. Functional OXT receptors (OXTRs) are found in bone cells in both human osteoblasts [114] and osteoclasts [115].
Oxt–/– and Oxtr–/– male and female mice displayed reduced bone mass with decreased bone formation. OXT treatment in cell culture stimulated osteoblast differentiation by upregulating Bmp2, Schnurri-2 and -3, osterix, and Atf-4 [116]. It also stimulated osteoclast formation by activating NFκB and mitogen-activated protein kinase (MAPK) signaling and indirectly through RANKL. Osteoclast-driven bone resorption was counteracted through cytosolic Ca2+ releases and nitric oxide (NO) synthesis, resulting in net anabolic effect [116]. In line with the findings, OXT decreased RANKL levels and increased OPG levels resulting in reduction in RANKL/OPG levels favoring net bone formation [117]. Osteoblast-specific Oxt–/–mice showed low bone mass, and osteoclast-specific Oxt–/– developed high bone mass, again confirming a stimulatory effect of OXT on osteoblastic bone formation and on osteoclast differentiation [118].
We believe that OXT might play a role in skeletal mobilization through increased osteoclast formation during pregnancy and lactation [119]. Oxt–/– pups showed hypomineralized skeleton and Oxt–/– mom displayed reduced bone formation markers [119]. However, interestingly, pregnant, and lactating mice lacking OXTRs in osteoblasts showed higher bone mass, suggesting elevated OXT inhibits bone resorption to keep a balance against excessive bone loss during pregnancy and lactation (Fig. 4) [118].
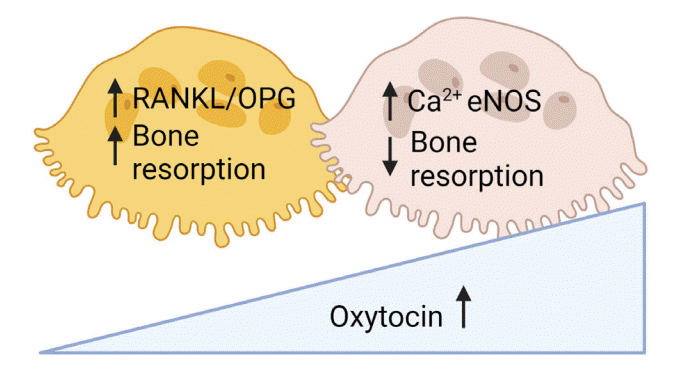 | Fig. 4.Oxytocin increases bone turnover by increasing osteoblastic bone formation and osteoclastic bone resorption with net anabolic effect. However, high levels of oxytocin upregulates endothelial nitric oxide synthase (eNOS) and inhibits osteoclastic bone resorption to counteract rapid bone loss in maternal skeleton during pregnancy and lactation. RANKL, receptor activator of NF-κB-ligand; OPG, osteoprotegerin.
|
Go to :
VASOPRESSIN AND BONE
Arginine vasopressin (AVP), a key regulator of serum osmolality and fluid status, has also been implicated in bone remodeling. AVPR-1a and -2a are expressed in the osteoblasts and osteoclasts [120]. Vasopressin-deficient (AVPR-1a) mice displayed high bone mass due to increased bone formation and reduced bone resorption [29,120]. In addition, AVP administration to mice reduced osteoblast formation and increased osteoclast formation. On the other hand, AVPR-1a antagonist (SR49059) increased bone mass by promoting osteoblastogenesis and inhibiting osteoclast-driven bone resorption, together suggesting that vasopressin negatively regulates skeletal remodeling [120]. In contrast, AVPR-2a does not seem to have any skeletal effect as AVPR-2a inhibitor tolvaptan did not show any skeletal phenotype [29]. Our finding might explain the bone loss and high risk of fracture in patients with chronic hyponatremia that is often accompanied by high vasopressin levels.
Go to :
CONCLUSIONS
The discovery of independent skeletal effects of pituitary hormones highlights the power of integrative bone physiology as it relates to the remodeling of bone and maintenance of its integrity. Bone interacts with not only closely situated organs—bone marrow, fat and muscle—but also with remote organs, such as the pancreas, brain, and kidney. The skeletal role of FSH explains, at least in part, the natural course of bone changes during the perimenopausal transition, together with its expanding function in the pathogenesis of obesity [14] and spikes of cognitive decline during this phase in a woman’s life [13]. Thus, we propose that FSH is an important aging hormone. Furthermore, the opposing skeletal actions of pituitary hormone and the induced master hormones suggest mutually compensatory effects to regulate skeletal homeostasis precisely. For example, hyperthyroid bone loss (low TSH and high T4/T3) could be counteracted by T4/T3-induced TSH-βv secretion from immune cells. In addition, effects of OXT and prolactin on the skeleton mark a potential role for the hormones during pregnancy and lactation. Likewise, the skeletal role of vasopressin suggests that sodium balance in the skeleton, which has not been studied well, also plays an important role in bone remodeling. In all, the exciting new out-of-the-box discoveries not only offer better understanding of bone biology, but also unmask potential therapeutic targets for osteoporosis.
Go to :
 XML Download
XML Download