

 PDF
PDF Citation
Citation Print
Print



INTRODUCTION
Carney complex (CNC) is a rare multiple neoplasia syndrome characterized by multiple endocrine and nonendocrine diseases, such as primary pigmented nodular adrenal disease (PPNAD), acromegaly, thyroid carcinoma, and spotty skin pigmentation [1,2]. A confirmed diagnosis of CNC is made in cases with two or more of these characteristic diseases or with one of these diseases and a family history of a first-degree relative or based on genetic testing results [2].
The protein kinase A regulatory subunit 1 alpha (PRKAR1A) gene, which is located in 17q24, has been implicated in CNC pathogenesis [3]. The PRKAR1A gene encodes a regulatory subunit of protein kinase A (PKA) and dissociates from the catalytic subunit when cyclic adenosine monophosphate (cAMP) binds [4]. Loss-of-function variants of PRKAR1A lead to uncontrolled activation of the cAMP/PKA signaling pathway, which causes CNC [5]. The PRKAR1A variant shows an autosomal dominant inheritance or arises sporadically. To date, more than 125 variants of PRKAR1A have been reported [2,6].
Here, we present a novel missense variant of PRKAR1A with uncertain significance identified in a family. We aimed to conduct in vitro functional studies to verify its role in causing CNC. This study was approved by the Institute Review Board of Seoul National University Hospital (IRB No. 2204-048-1314).
RESULTS
Clinical description
A 21-year-old woman visited the endocrinology department for hirsutism of her chin and face. Laboratory tests showed insulinlike growth factor-I (IGF-I) and growth hormone (GH) levels to be 893 ng/mL (reference, 219 to 644) and 12.06 ng/mL (reference, 0.3 to 8.7), respectively. In the 75 g oral glucose tolerance test, the nadir GH level was 2.42 ng/mL. Pituitary magnetic resonance imaging showed a 1.2 cm-sized pituitary adenoma (Supplemental Fig. S1). The patient was diagnosed with acromegaly and underwent transsphenoidal surgery and her IGF-I level normalized. The patient was lost to follow-up after surgery.
Two years later, she revisited the hospital due to headaches, weight gain, and high blood pressure. Her IGF-I and GH levels were within the normal range. Examinations showed midnight cortisol, adrenocorticotropic hormone (ACTH), and 24-hour urinary free cortisol levels to be 9.0 μg/dL, 1.0 pg/mL, and 106.8 μg/day (reference, 13.8 to 75.4), respectively. Her serum cortisol level after overnight dexamethasone suppression was 10.1 μg/dL. Adrenal computed tomography revealed multiple small nodules in both adrenal glands (Fig. 1A). We planned left unilateral adrenalectomy due to her young age and mild hypercortisolism. Pigmented micronodules were observed throughout the cortex of the adrenal gland in the patient upon adrenalectomy (Fig. 1B). She was diagnosed with CNC based on typical spotty skin pigmentation and two endocrine disorders, acromegaly and PPNAD.
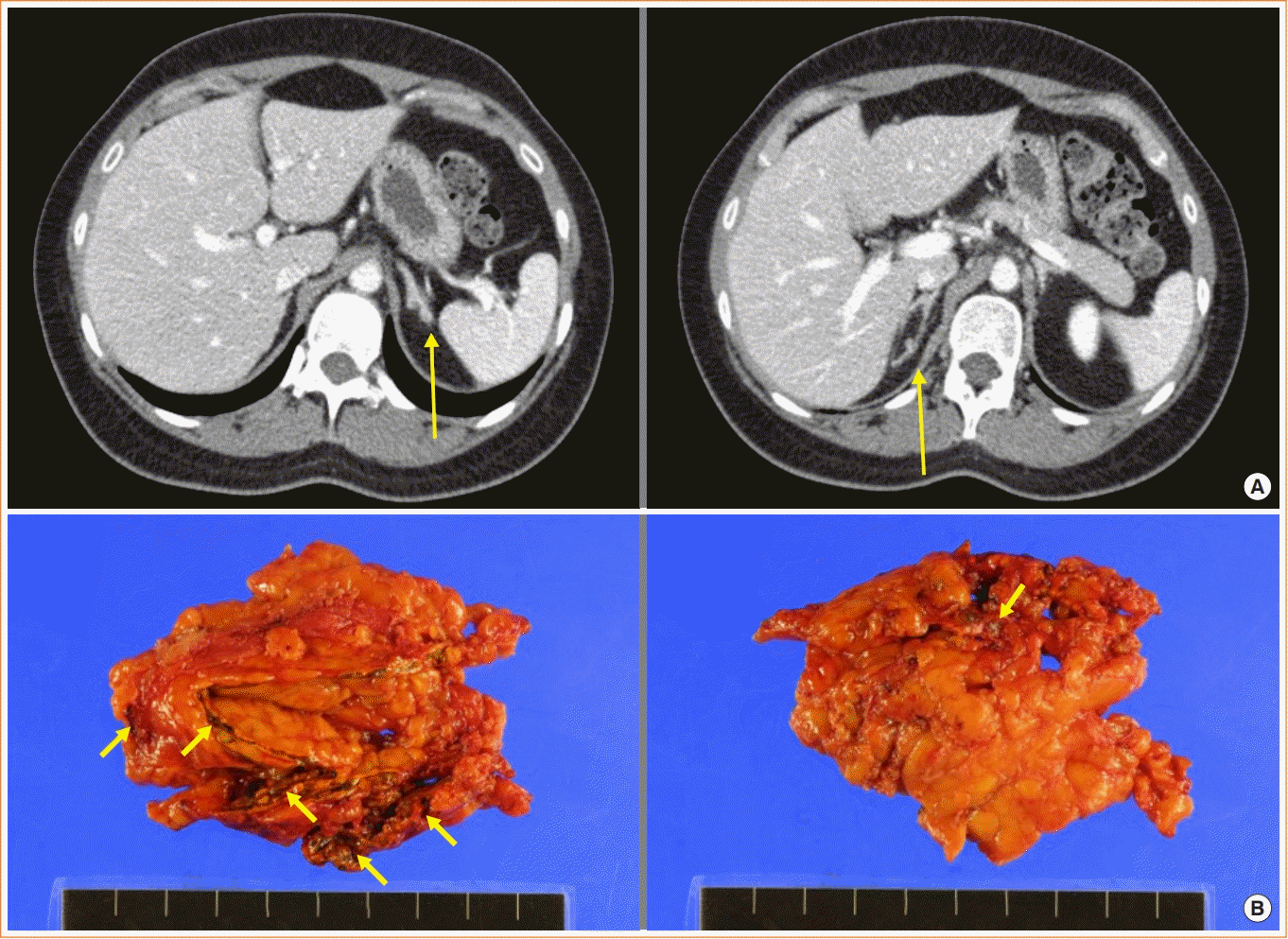
Fig. 1.
The patient’s adrenal gland. (A) Adrenal computed tomography was performed as cortisol level was elevated in the examination, considering secondary hypertension. Adrenal computed tomography revealed multiple small nodules in both the adrenal glands (arrows). (B) Pigmented micronodules were observed throughout the cortex of the adrenal gland in the patient who underwent adrenalectomy (arrows).
![]()
Intensive family history for the patient was recorded. Her mother had a history of adrenal Cushing’s syndrome and acromegaly. In addition, her two uncles (II.6, II.8) and male cousins (III.5, III.6) showed acromegaly (Fig. 2A). These findings corroborated the diagnosis of CNC and the autosomal dominant inheritance. Molecular testing and genetic counseling were recommended to the family.
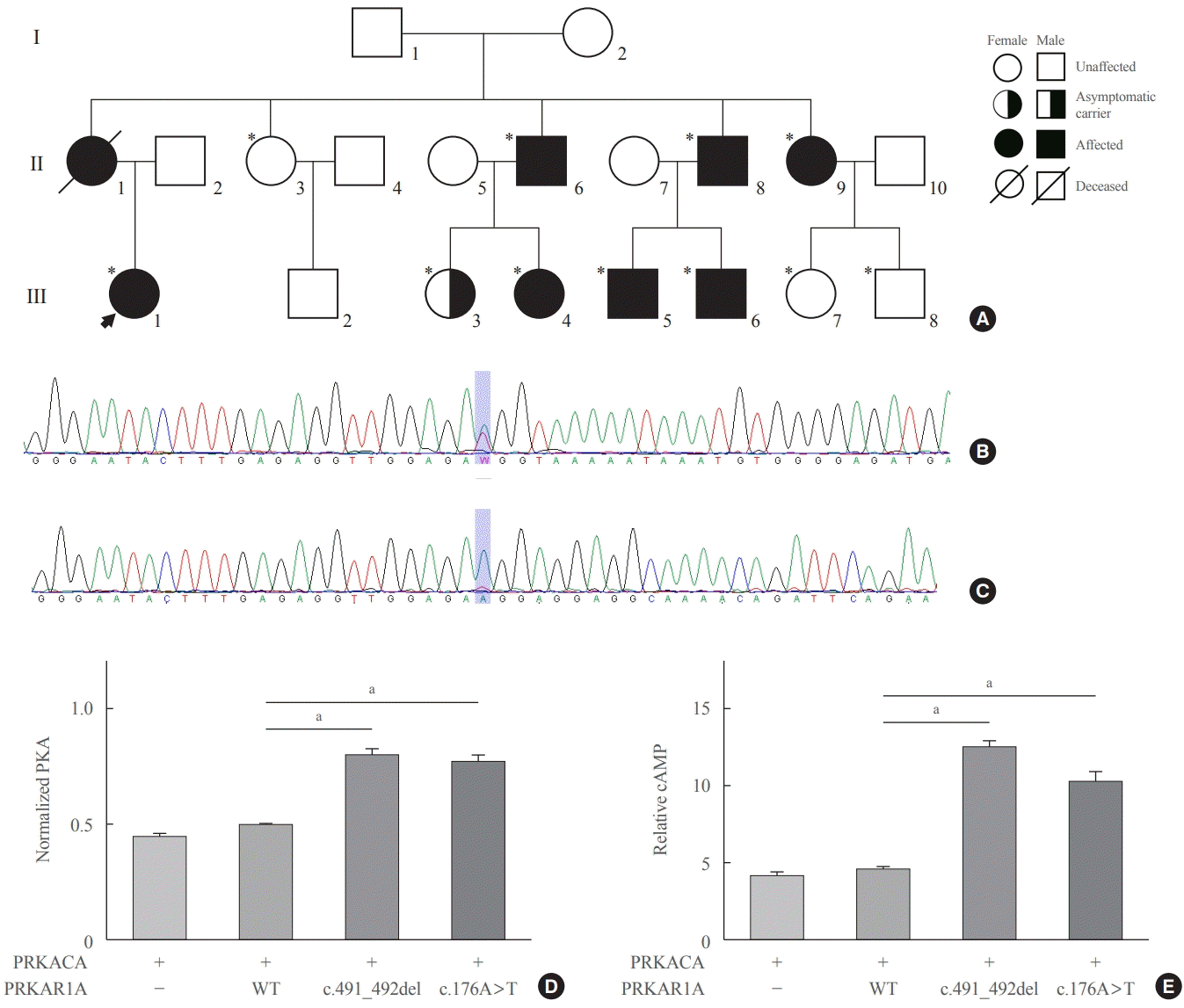
Fig. 2.
The patient’s pedigree analysis, sequencing analysis of protein kinase A regulatory subunit 1 alpha (PRKAR1A), and the protein kinase A (PKA) activity and cyclic adenosine monophosphate (cAMP) levels in PRKAR1A mutants. (A) Family tree of the patient diagnosed with the Carney complex. The patient’s mother had acromegaly and adrenal Cushing’s syndrome and died of colon cancer. Both her uncles were treated for acromegaly. Genetic testing confirmed the same variant in all of her mother’s brothers, one sister, and four symptomatic cousins (*). Two cousins (III.3 and III.8) are asymptomatic carriers. Family members who underwent genetic testing for the variant of PRKAR1A. (B) DNA sequencing result showing a novel missense variant, NM_002734.5(PRKAR1A): c.176A>T. (C) RNA sequencing analysis showing the absence of the variant. (D) Relative PKA activity was significantly higher in both a previously known mutant (c.491_492del) and the new mutant found in the patient of this study (c.176A>T) than in the wild type (WT), and there was no significant difference in the relative PKA activity between the c.491_492del and c.176A>T mutants. (E) Relative cAMP level in both the c.491_492del and c.176A>T mutants was significantly higher than that in the WT. PRKACA, protein kinase cAMP-activated catalytic subunit alpha. aP<0.001.
![]()
Molecular analysis of PRKAR1A
Sanger sequencing of the PRKAR1A gene using genomic DNA with the primers (Supplemental Table S1) was performed to diagnose CNC. Genetic testing revealed a novel heterozygous variant of PRKAR1A, NM_002734.5:c.176A>T, located at the 3′ margin of exon 2 (Fig. 2B). This variant has not been reported in the public or the variant database and was predicted to be deleterious by MutationTaster [7] and Sorting Intolerant From Tolerant (SIFT) [8] but benign by PolyPhen-2 [9]. Therefore, it was classified as a variant of uncertain significance [10].
Meanwhile, splicing-site prediction of the variant was as high as 0.9999 (adaptive boosting score) and 0.9959 (random forests score) using dbscSNV [11]; leading to the possibility of aberrant splicing. We performed reverse transcription polymerase chain reaction to assess the splicing effect, and the same variant was not identified in the RNA sample from the fresh frozen tissue of the adenoma (Fig. 2C), suggesting aberrant splicing and nonsense-mediated mRNA decay (NMD).
Genetic testing of the variant was performed on her relatives. The same variant was identified in her uncles (II.6, II.8) and male cousins (III.5, III.6) diagnosed with acromegaly. Additionally, her aunt (II.9) and female cousin (III.4), who had a history of breast fibroadenoma, had the same variant. Two other cousins (III.3, III.8) also had the same variant, but they were asymptomatic (Fig. 2A).
Functional analysis of the missense variant
Protein kinase activity and cAMP levels were compared between the wild type PRKAR1A, a previously known mutant (c.491_492del), and the new mutant found in this patient (c.176A>T) which were generated by site-directed mutagenesis using the primers (Supplemental Table S2). Compared with the wild type, the relative protein kinase activity was significantly higher in both mutants, and there was no significant difference in the relative protein kinase activity between both the mutants (0.49±0.04 vs. 0.80±0.08 vs. 0.77±0.07, P<0.001) (Fig. 2D). The relative cAMP level was significantly lower in the new mutant than that in the known mutant (10.14±2.47 vs. 12.45±1.48, P<0.001) (Fig. 2E), but the relative cAMP level in both mutants was significantly higher than that in the wild type. Collectively, our observations suggested that the variant caused augmentation of PKA activity by leveraging the haploinsufficiency of PRKAR1A.
DISCUSSION
We report a patient with CNC who underwent surgery for acromegaly and was later diagnosed with adrenal Cushing’s syndrome due to PPNAD. In this patient, a novel variant of PRKAR1A was found, which increased PKA activity and cAMP levels compared to the wild type. The patient had a family history of adrenal Cushing’s syndrome and acromegaly, and the same PRKAR1A variant was found in the family.
A retrospective study of 353 patients with PRKAR1A variants or with CNC or PPNAD was published, highlighting that 60% of the patients presented with PPNAD, with the median age of its diagnosis being 34 years. In addition, cardiac myxoma was found in 32% of the patients and was diagnosed at a median age of 50 years [12]. Subsequently, a prospective study was published in which 70 patients with PRKAR1A variants or patients diagnosed with CNC or PPNAD [13]. The onset age of the typical diseases of CNC is different, with the possibility of new diseases occurring with follow-up. Hence, patients with CNC should be monitored regularly with a detailed examination of all the clinical manifestations of CNC [2].
Among the reported pathogenic variants of PRKAR1A, most are frameshifts, splice sites, insertions, or nonsense variants that cause premature stop codons and undergo NMD [14]. To date, 14 missense variants have been reported, only two of which have been reported to lead to NMD [15]. In this report, a novel missense variant was identified. We considered that this variant possibly caused aberrant splicing due to the location of the variant. RNA analysis showed the degradation of the mutant mRNA. In addition, a functional study showed that this variant caused an increase in PKA activity, indicating that all PRKAR1A variants should be considered, particularly in patients with CNC. PRKAR1A variants have an almost complete penetrance; therefore, family members of CNC patients should be recommended for genetic testing [16].
In conclusion, we identified a novel missense PRKAR1A variant in a patient with CNC that caused NMD and PRKAR1A haploinsufficiency. Through a functional study, we confirmed the pathogenic role of the novel missense PRKAR1A variant. This study contributes to the understanding of PRKAR1A-associated CNC.
 XML Download
XML Download