

 PDF
PDF Citation
Citation Print
Print



INTRODUCTION
Vascular smooth muscle cells (VSMCs) are abundant in the medial layer of arteries and help to maintain vessel tone, the bloodstream, and blood pressure [1]. Abnormal proliferation and migration of VSMCs play an essential role in occlusive vascular diseases, including atherosclerosis and intimal hyperplasia induced by vascular injury [2]. In response to stimuli that directly cause vascular injuries, such as angioplasty, stent insertion, and coronary artery bypass grafting, intimal hyperplasia in arteries is promoted, and injured arteries are prone to develop vascular restenosis [3,4]. Given that VSMC proliferation and migration have a pivotal role in the pathogenesis of occlusive vascular diseases, accumulating studies have focused on discovering pharmacological molecules that inhibit VSMC proliferation and migration [2,5,6].
Rapidly proliferating VSMCs require not only increased glycolysis but also enhanced oxidative phosphorylation to meet their increased demands for energy and anabolic precursors [7-9]. Platelet-derived growth factor (PDGF)-stimulated VSMCs exhibit increases in basal respiration, maximal respiratory capacity, and adenosine triphosphate (ATP)-linked respiration in mitochondrial respiration analysis [10-12], whereas proliferation of VSMCs is suppressed when mitochondrial oxidative phosphorylation is inhibited [13-16]. Given that anaplerosis of the citric acid cycle, which is maintained mainly by glutaminolysis, is critical in proliferating cells [17], blockade of glutaminolysis in growth factor-stimulated VSMCs is considered a therapeutic strategy to prevent VSMC proliferation and neointima formation [8,10,18].
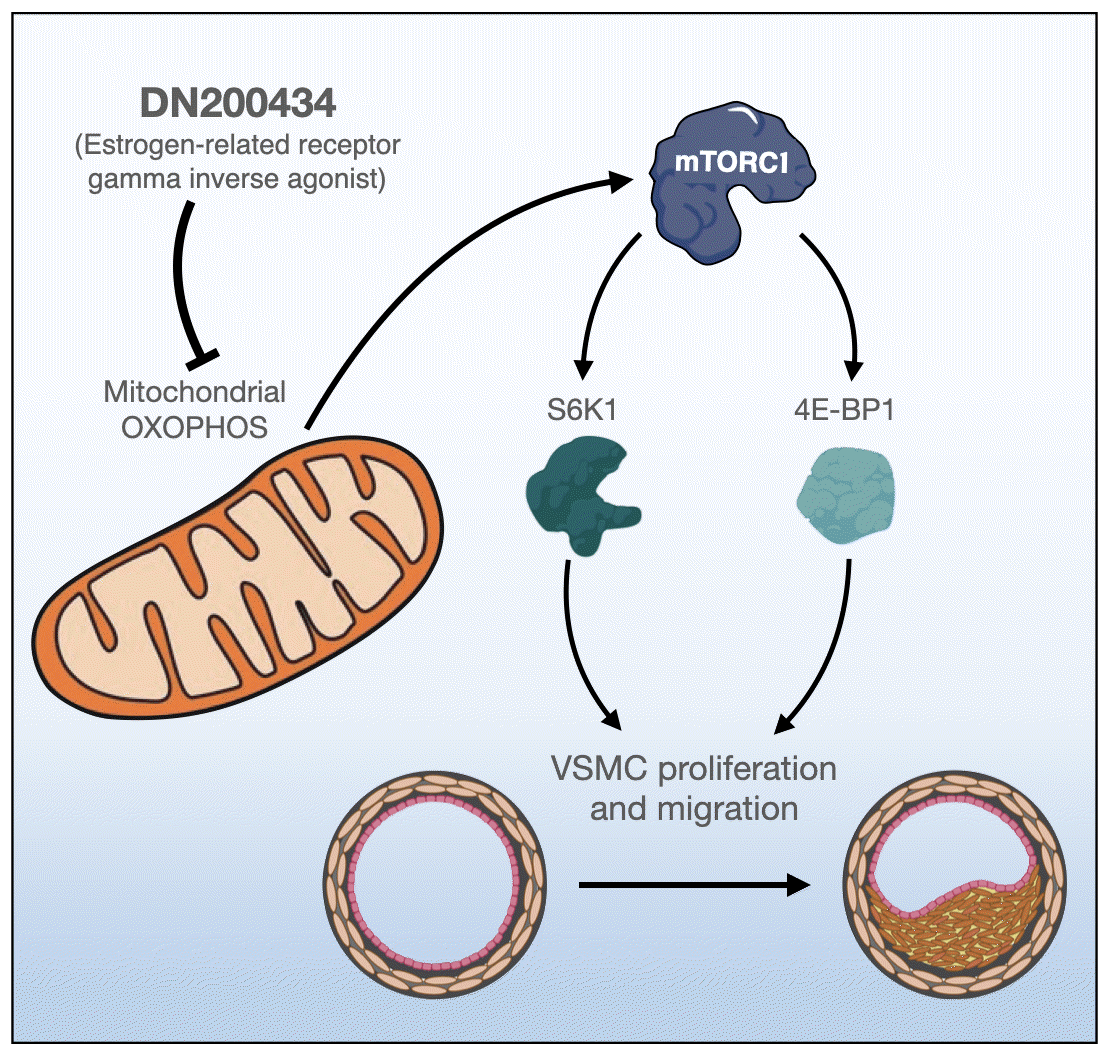
Estrogen-related receptor gamma (ERRγ) is expressed in metabolically active organs such as the heart, kidney, pancreas, and skeletal muscle [19]. It positively regulates mitochondrial bioenergetics, oxidative phosphorylation, and cellular energy homeostasis [20,21]. DN200434, an orally available ERRγ inverse agonist, is a newly developed compound [22]. It effectively enhances radioiodine avidity of anaplastic thyroid carcinoma tumors by upregulating the sodium iodide symporter [22]. However, the effects of DN200434 on proliferation of VSMCs and restenosis remain to be elucidated.
In this study, we investigated whether DN200434 inhibits proliferation and migration of VSMCs and mitochondrial oxidative phosphorylation, and subsequently suppresses neointima formation in a mouse carotid artery ligation model.
METHODS
Cell culture
Rat aortic VSMCs were prepared as described previously [23]. Briefly, rat aortic smooth muscle cells were isolated from 4-week-old male Sprague-Dawley rats (90 to 100 g) and cultured in plastic culture dishes containing low-glucose Dulbecco’s modified Eagle’s medium (Hyclone, Logan, UT, USA) supplemented with 20% fetal bovine serum (FBS, Hyclone) for 2 weeks at 37°C in 5% CO2. The medium was changed daily. Cells at passage 4 to 9 were used in experiments.
Cell counting
Primary VSMCs were cultured for 18 hours under serumstarved conditions and then incubated for 24 hours in the presence or absence of 10% FBS or platelet-derived growth factor subunit B (PDGF-BB, 20 ng/mL) with or without 10 μM DN200434 (Daegu Gyeongbuk Medical Innovation Foundation, Daegu, Korea). Hemocytometer-based cell counting was performed using trypan blue solution.
Western blot analysis
Western blot analysis was performed as previously described [10]. Protein samples were loaded on sodium dodecyl sulfate–polyacrylamide gel electrophoresis (SDS-PAGE) gels and transferred from gels to polyvinylidene difluoride membranes (Millipore, Danvers, MA, USA). The membranes were washed and blocked with 5% skim milk prepared in Tris-buffered saline containing 0.1% Tween 20 (TBST) and incubated overnight at 4°C with primary antibodies against 70 kDa ribosomal protein S6 kinase (p70S6K), phospho-p70S6K (T389), eukaryotic initiation factor 4E-binding protein 1 (4E-BP1), phospho-4E-BP1 (T37/46), proliferating cell nuclear antigen (PCNA), and cyclin D1 (Cell Signaling Technology, Danvers, MA, USA), as well as β-actin (Sigma, St. Louis, MO, USA). The blots were rinsed with TBST and incubated with horseradish peroxidase (HRP)-conjugated secondary antibodies (Santa Cruz Biotechnology, Dallas, TX, USA). HRP was detected using enhanced chemiluminescence reagent (BioNote, Hwaseong, Korea).
Migration assay
For the wound healing assay, VSMCs (1×105 cells per well) were plated into a 6-well plate and serum-starved for 18 hours. An artificial wound (scratch) was generated using a 200 μL pipette tip. Cells were incubated with or without DN200434 (10 μM) for 24 hours in the presence or absence of 10% FBS or PDGF-BB (20 ng/mL). When the wound had closed, cells were fixed in 4% paraformaldehyde and stained with 0.05% crystal violet (Sigma). For the Transwell migration assay, VSMCs (1×104 cells per well) were seeded onto the microporous membrane (8.0 μm) in the upper chamber of the Transwell (Corning Incorporated, Kennebunk, ME, USA). Cells were serum-starved for 18 hours and then incubated with or without DN200434 for 24 hours in the presence or absence of 20% FBS or PDGF-BB (20 ng/mL). Non-migrated cells in the upper chamber were removed using a cotton swab. Cells that had migrated through the membrane to the lower chamber were fixed with methanol and stained with 0.05% crystal violet. Quantification of transwell migration and wound area were performed using Image J software (National Institutes of Health, Bethesda, MD, USA).
Immunohistochemistry
Immunohistochemistry (IHC) was performed on formalinfixed, paraffin-embedded tissue sections, as previously described [10]. The sections were incubated with an anti-PCNA (1:1,500, Cell Signaling Technology) or anti-phospho-4E-BP1 (T37/46) (1:50, Cell Signaling Technology) antibody, and then with a HRP-conjugated secondary antibody, followed by staining with diaminobenzidine (liquid DAB+Substrate Chromogen System, Dako, Carpinteria, CA, USA) and counterstaining with Mayer’s hematoxylin (Merck Inc., Darmstadt, Germany) at room temperature. Images were obtained using a light microscope and the cellSens Entry 1.9 imaging system (Olympus Corporation, Tokyo, Japan). IHC images were quantified using ImageJ software (National Institutes of Health).
Flow cytometric analysis
The cell cycle of VSMCs treated with 10% FBS or PDGF-BB (20 ng/mL) with or without 10 μM DN200434 was analyzed by flow cytometry. Cells were serum-starved for 6 hours to synchronize them in G1 phase and then incubated with or without DN200434 for 12 hours in the presence or absence of 10% FBS or PDGF-BB (20 ng/mL). Thereafter, cells were washed with ice-cold phosphate-buffered saline, fixed for 2 hours in cold (–20°C) 70% ethanol, and stained for 30 minutes with propidium iodide (PI)/RNase Staining Buffer (BD Biosciences, San Diego, CA, USA) in the dark. Fluorescence emitted by PI-DNA complexes was measured using an Accuri C6 flow cytometer (BD Biosciences).
Measurement of the oxygen consumption rate
The oxygen consumption rate (OCR) of VSMCs was measured using a XF-24 Seahorse Extracellular Flux Analyzer (Seahorse Bioscience, North Billerica, MA, USA). VSMCs were starved for 18 hours and incubated with 10% FBS or PDGF-BB (20 ng/mL) with or without DN200434 (10 μM) for 24 hours. XF Calibrant solution was added to each well of a Seahorse XF-24 plate and incubated at 37°C in a non-CO2 incubator for 1 hour. To investigate changes in cell respiration, 1 μM oligomycin (Sigma), 2 μM carbonyl cyanide 3‑chlorophenylhydrazone (Sigma), and 1 μM rotenone (Sigma) were injected at the indicated time points during the measurement. OCR data were obtained automatically following the manufacturer’s protocol.
Murine model of carotid artery ligation
To investigate the effect of DN200434 on neointima formation in vivo, the left common carotid artery of male C57BL/6J mice was ligated as previously described [24]. Briefly, the left common carotid artery proximal to the distal bifurcation was ligated with a 5.0 silk suture, and mice were maintained for 28 days. DN200434 (100 mg/kg/day via oral gavage, 5 days per week for 4 weeks) was orally administered prior to scheduled sacrifice. Arteries proximal to the ligation site were analyzed using hematoxylin and eosin (H&E) and an Elastic van Gieson (EVG) staining kit (Abcam, Cambridge, UK). The cross-sectional intimal and medial areas were measured using ImageJ software (National Institutes of Health). The ratio of the intimal area to the medial area was calculated from the mean of these measurements.
Ethical statement
All animal procedures were approved by the Institutional Animal Care and Use Committee of Kyungpook National University (KNU2020-0145).
Statistical analysis
All values in graphs represent the mean±standard error of the mean. Statistical analysis to assess differences between groups was performed using a one-way analysis of variance and Dunnett’s post-test (GraphPad Prism 8.0, GraphPad, San Diego, CA, USA). P<0.05 was considered to indicate a statistically significant difference.
RESULTS
DN200434 inhibits FBS- or PDGF-stimulated VSMC proliferation and cell cycle progression
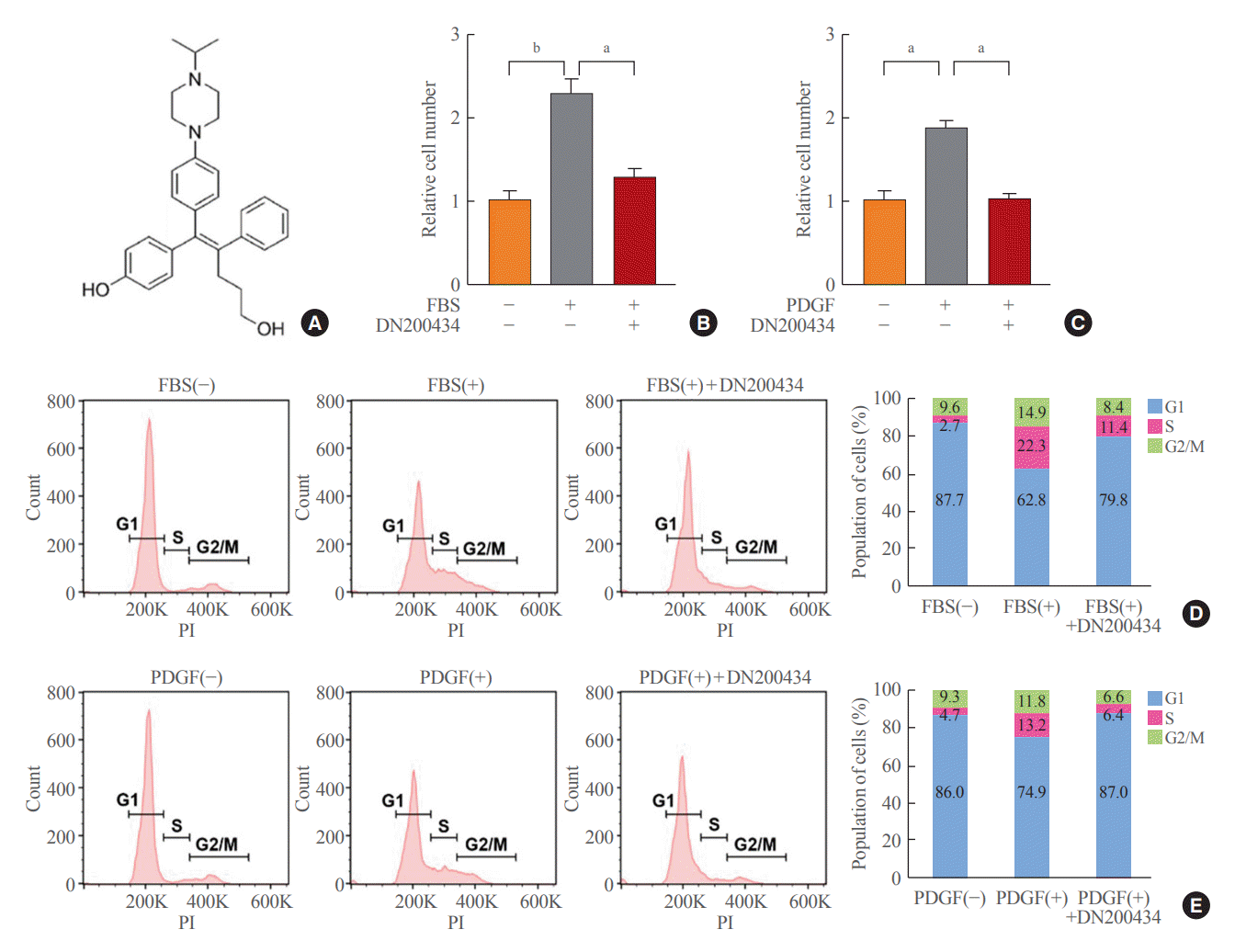
The chemical structure of DN200434 is shown in Fig. 1A. DN20-0434 significantly suppressed FBS- or PDGF-stimulated VSMC proliferation (Fig. 1B, C). In addition, flow cytometric analysis of the cell cycle showed that DN200434 attenuated FBS- or PDGF-stimulated progression from G1 to S phase (Fig. 1D, E).
DN200434 attenuates mitochondrial respiration in FBSor PDGF-stimulated VSMCs
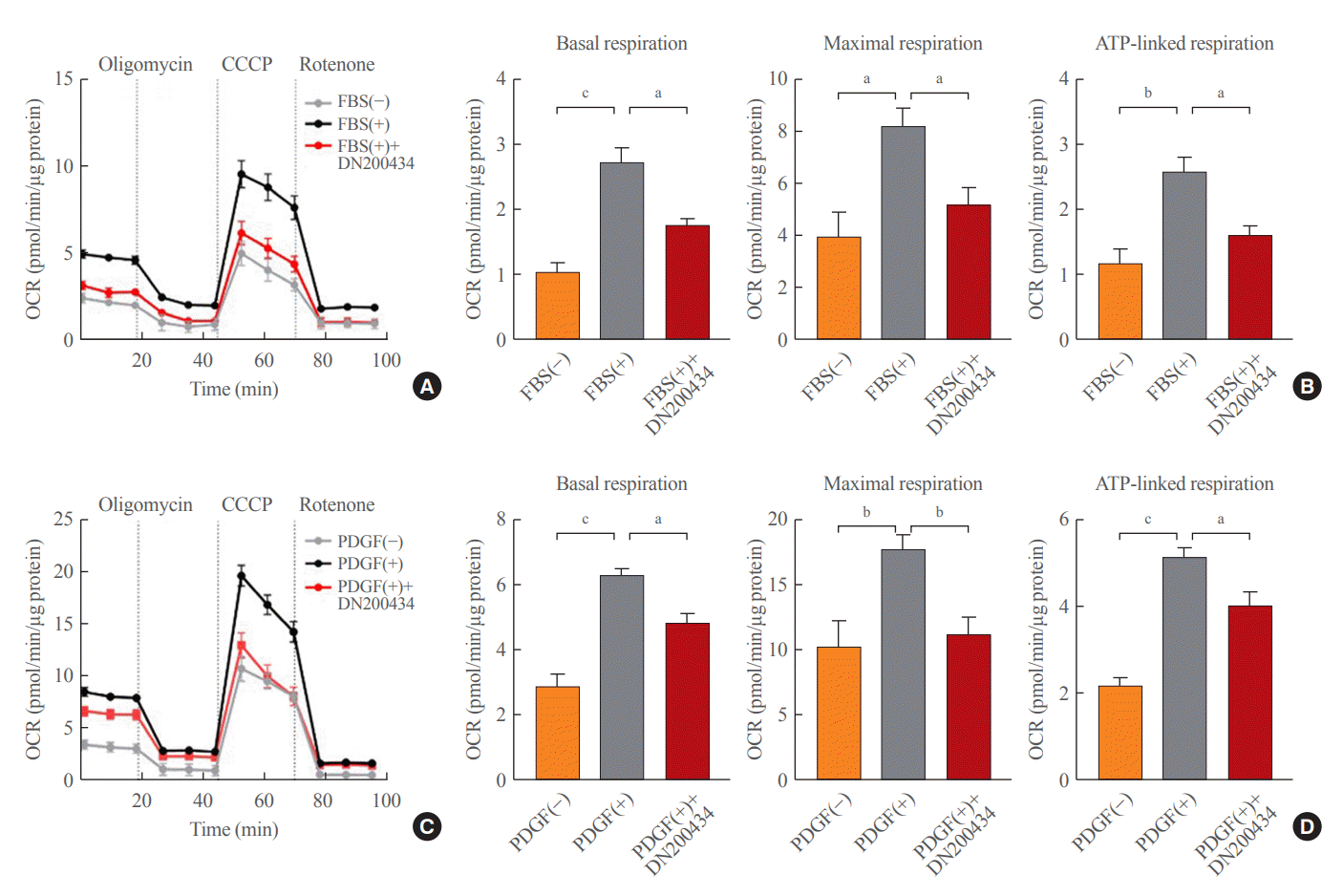
Given that oxidative phosphorylation has a role in VSMC proliferation [8,10], we investigated the effects of DN200434 on the OCR of FBS- or PDGF-stimulated VSMCs using an XF analyzer. Consistent with a previous report [8], FBS or PDGF stimulation significantly increased the major parameters of mitochondrial functions, including basal and maximal respiration and ATP-linked respiration, in VSMCs, and these increases were markedly attenuated by DN200434 (Fig. 2).
DN200434 inhibits mTORC1 activity and attenuates cell proliferation and migration in FBS- or PDGF-stimulated VSMCs
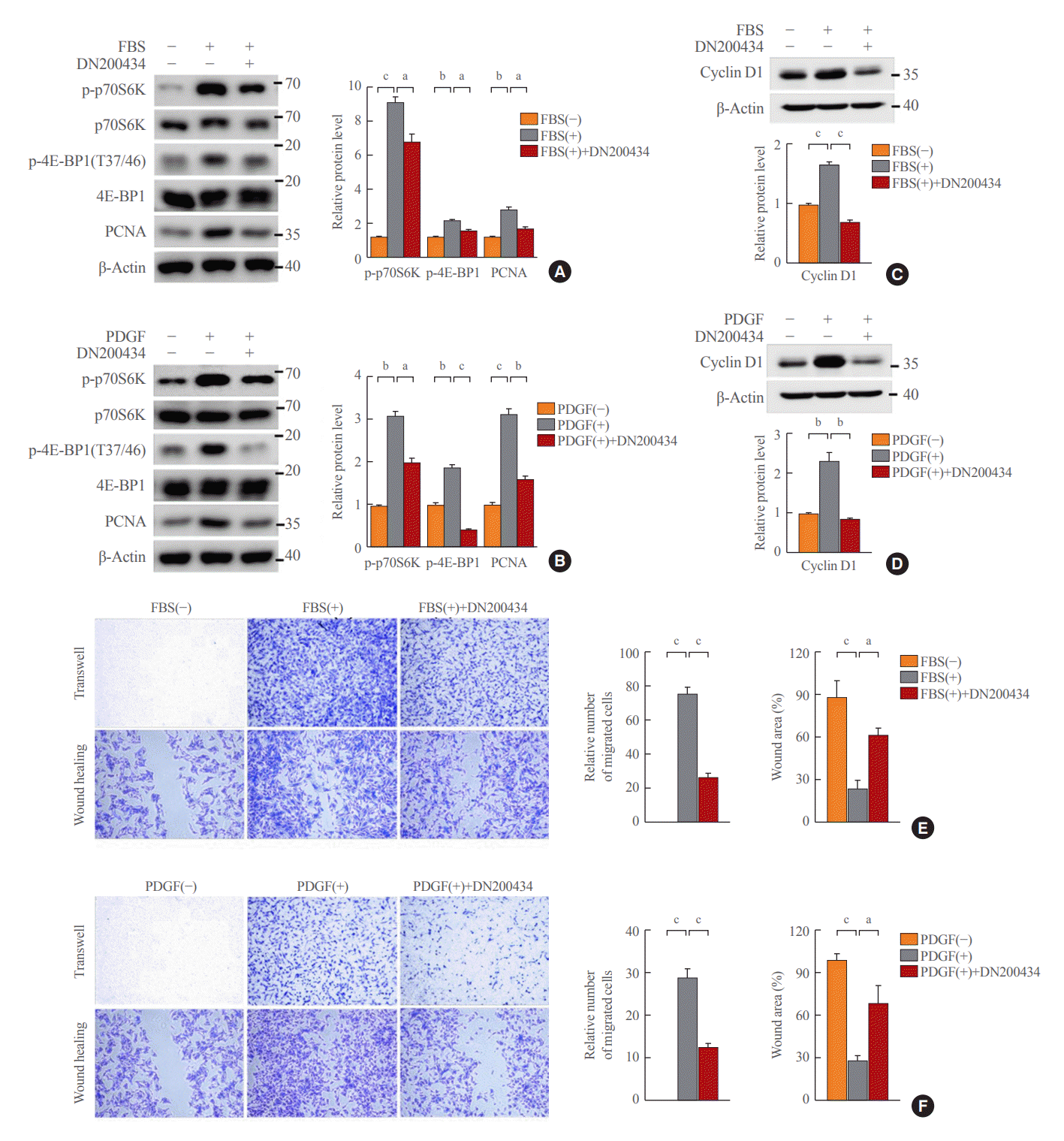
Given that decreased ATP production due to reduced mitochondrial respiration activity inhibits mammalian target of rapamycin complex 1 (mTORC1) activity [25], we investigated the effect of DN200434 on mTORC1 activity in VSMCs. DN200434 inhibited the FBS- or PDGF-induced increases in phosphorylation of p70S6K and 4E-BP1, which are two major downstream targets of mTORC1 (Fig. 3A, B). Furthermore, DN200434 markedly reduced the PCNA level in response to FBS or PDGF stimulation (Fig. 3A, B). Expression of cyclin D1, which controls the transition from G1 to S phase, was increased in FBS- or PDGF-stimulated VSMCs, and these increases were attenuated by DN200434 (Fig. 3C, D). Furthermore, the wound healing and Transwell chamber assays showed that DN200434 attenuated FBS- or PDGF-stimulated migration of VSMCs (Fig. 3E, F).
DN200434 reduces carotid artery ligation-induced neointimal hyperplasia
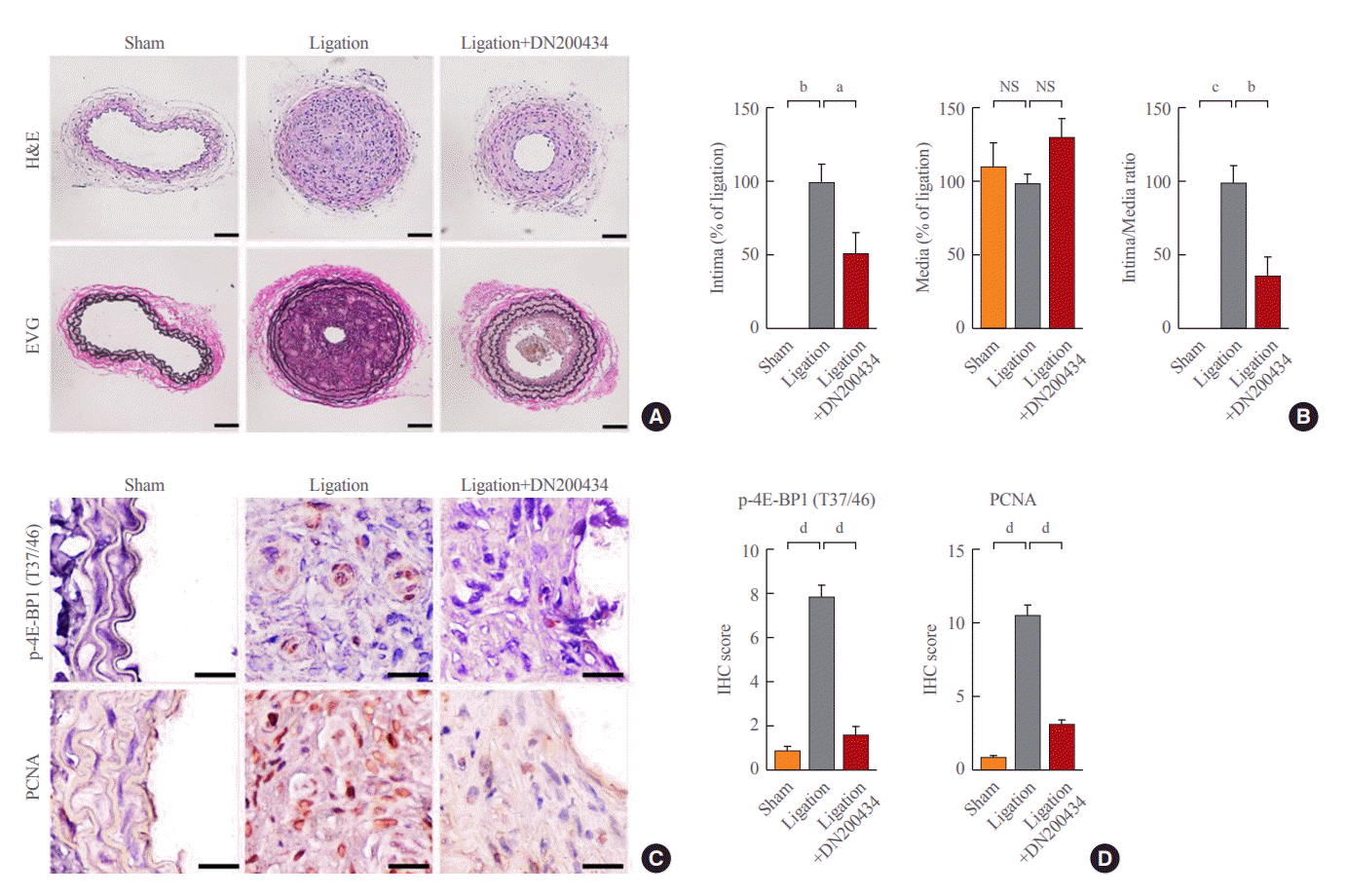
Finally, we investigated the effect of DN200434 on neointima formation using a mouse carotid artery ligation model. H&E and EVG staining of arteries showed severe neointima formation at 4 weeks after carotid artery ligation. Ligation-induced neointimal hyperplasia and the increase in the ratio of intimal area to medial area were significantly attenuated in the DN20-0434-treated group compared with the ligation only group (Fig. 4A, B). Consistent with the effects of DN200434 in vitro, immunohistochemical staining of the carotid artery showed that DN200434 attenuated the increased expression of phospho-4E-BP1 (T37/46) and PCNA in ligation-induced neointimal hyperplasia (Fig. 4C, D).
DISCUSSION
In the present study, DN200434 inhibited growth factor-stimulated proliferation, migration, and cell cycle progression of VSMCs by reducing mitochondrial respiration and mTORC1 activity. Moreover, DN200434 significantly attenuated ligationinduced neointimal formation in mice.
Mitochondrial oxidative phosphorylation is essential in rapidly proliferating VSMCs to meet the increased demands for energy and biomass [26-28]. Our previous study showed that suppression of mitochondrial oxygen consumption via inhibition of glutamine metabolism significantly attenuates VSCM proliferation and neointima formation [8,10]. A growing body of evidence suggests that ERRγ is associated with expression of genes involved in mitochondrial metabolism, including the tricarboxylic acid cycle, fatty acid oxidation, glutamine metabolism, and mitochondrial oxidative respiration [29,30]. A recent study reported that the ERRγ inverse agonist GSK5182 reduces glutamine metabolism and represses glutamine oxidative metabolism in cells exposed to oxidative stress [30]. Consistent with these previous studies, we observed that DN200434 significantly inhibited mitochondrial oxidative phosphorylation in growth factor-stimulated VSMCs.
Impairment of mitochondrial function negatively regulates mTORC1 activity [8,10]; therefore, reduction of mitochondrial oxidative phosphorylation by DN200434 in VSMCs is critical for suppression of mTORC1 activity. mTORC1 plays a pivotal role in cell growth and proliferation, and activation of mTORC1 is induced by several factors, such as growth factors, ATP, amino acids, and mechanical stimuli [31-36]. mTORC1 activity contributes to aberrant VSMC proliferation and migration, which have a pivotal role in development of atherosclerosis and formation of atherosclerotic plaques; thus, inhibition of mTORC1 is an effective strategy to reduce plaque size and complexity [36,37]. Indeed, mTORC1 activity has been commonly targeted with rapamycin and its analogues to prevent instent restenosis and vascular graft stenosis in clinical practice [38,39]. Furthermore, an mTORC1 inhibitor can induce cell cycle arrest in G1 phase and thus is used in the clinical setting [38,39]. We previously demonstrated the anti-atherosclerotic effects of the glutamine transporter inhibitor V-9302 and the glutamine antagonist 6-diazo-5-oxo-L-norleucine (DON) by showing that they decrease mTORC1 activity and mitochondrial oxidative phosphorylation in VSMCs [8,10]. In the present study, we found that DN200434 abrogated growth factor-induced mTORC1 activity and proliferation and migration of VSMCs. Based on our findings that DN200434 decreased the cyclin D1 protein level and induced cell cycle arrest, we conclude that it inhibits proliferation, migration, and cell cycle progression of VSMCs by reducing growth factor-stimulated mTORC1 activity, and consequently has anti-atherosclerotic effects.
In summary, the results of this study demonstrate that DN200434 suppresses VSMC proliferation and migration by inhibiting mitochondrial oxidative phosphorylation and mTORC1 activity. Given that this newly developed compound can control proliferation of VSMCs, DN200434 is expected to prevent vascular occlusive diseases such as atherosclerosis and restenosis.
 XML Download
XML Download