

 PDF
PDF Citation
Citation Print
Print



Introduction
Alzheimer disease (AD), age-dependent dementia characterized by irreversible and progressive loss of memory and cognition, shows an approximately 11.3% prevalence in patients aged 65 years and older in the United States [1]. The prevalence of dementia was reported to be 10.2% in the Republic of Korea, of which approximately 74.5% were diagnosed with AD [2]. Both reports showed that the incidence of AD increases with age and that the prevalence of AD is expected to increase until 2050. The cause of AD is not completely understood [3], and its pathophysiology is associated with amyloid-beta (Aβ) and tau protein accumulation, glial dysfunction, neurodegeneration (loss of neuronal connections), and altered oscillatory network activity [1,4-6].
Approximately 70% of the risk of AD is believed to be inherited from, with many genes usually involved [7]. Glenner and Wong [8] first suggested a correlation between cerebrovascular Aβ protein and Down syndrome (trisomy 21), which is homologous to AD. In dominantly inherited AD, missense mutations in amyloid precursor protein (APP) or presenilin-1/-2 genes on chromosome 21 increase Aβ production. Nondominant AD increases Aβ levels in the brain via the failure of Aβ clearance. Both of these situations result in the accumulation of Aβ42 oligomers in limbic systems. Affluent diffuse Aβ plaques without neuritic dystrophy, microgliosis, astrocytosis, and tangle formation have been observed in people who died in their early to mid-teens because of familial AD [9]. Aβ42 oligomers, which have been isolated from late-onset AD brains, reduce synapse density, suppress prolonged potentiation, and reinforce prolonged synaptic depression in the rodent hippocampus [9], and intraventricular injection of Aβ42 oligomers damages memory in healthy mature rats [10].
Thus, Aβ could directly or indirectly injure synapses and induce neuritis [9]. Aβ42 oligomers in patients with AD could also induce tau phosphorylation, which is associated with an increase in neurofibrillary tangles and neurotoxicity [9,11]. Since the discovery of Aβ protein, the Aβ hypothesis [12,13] has become the dominant model of AD pathogenesis and is guiding the development of potential therapeutic strategies. Although it is unclear how Aβ accumulates in the central nervous system and subsequently initiates AD, the generation of Aβ may occur in the neuronal axonal membranes after APP-mediated axonal transport of β-secretase, γ-secretase, and presenilin-1 [14,15], thus forming senile plaques outside neurons [16,17].
According to the Aβ hypothesis, several strategies have been identified as possible interventions against Aβ [18], including inhibitors against β-secretase or γ-secretase, selective Aβ42-lowering agents, and immunotherapy against Aβ. The results of a few clinical trials with monoclonal antibodies to Aβ have suggested a significant cognitive decline in patients with mild, but not moderate AD [9], but most immunotherapies eventually failed in phase II (crenezumab and gantenerumab) or phase III (solanezumab, aducanumab, and bapineuzumab) clinical trials [19]. These failures of Aβ monoclonal antibodies imply the need for a new approach to treat patients with AD.
The ion channel hypothesis postulates that oligomers of soluble, nonfibrillar Aβ form membrane ion channels, allowing the unregulated calcium influx into neurons [20,21] that underlies the disrupted calcium ion homeostasis and apoptosis seen in AD [22]. Optogenetics is a neuromodulation method that uses a combination of genetic methods and optical instruments to allow light to modulate the specific molecular and cellular activities of individual neurons in living tissue [23-26]. In this review, we will discuss the historical applications of optogenetics to investigate the mechanisms and possible therapeutic strategies involved in AD based on the Aβ hypothesis.
Optogenetic technique as a new neuromodulatory method
After Crick [27] speculated the concept of using light to control neuronal activity in 1979, Callaway and Katz [28] used light to uncage glutamine in living brain slices. Zemelman et al. [29] developed a targeting method using light to control rhodopsin-sensitized neurons. Nagel et al. [30] first applied the optogenetic manipulation of cation-selective ion movement by expressing channelrhodopsin-2 in Xenopus laevis and mammalian cells. Boyden et al. [23] used channelrhodopsin-2 to control neuronal spiking and synaptic transmission.
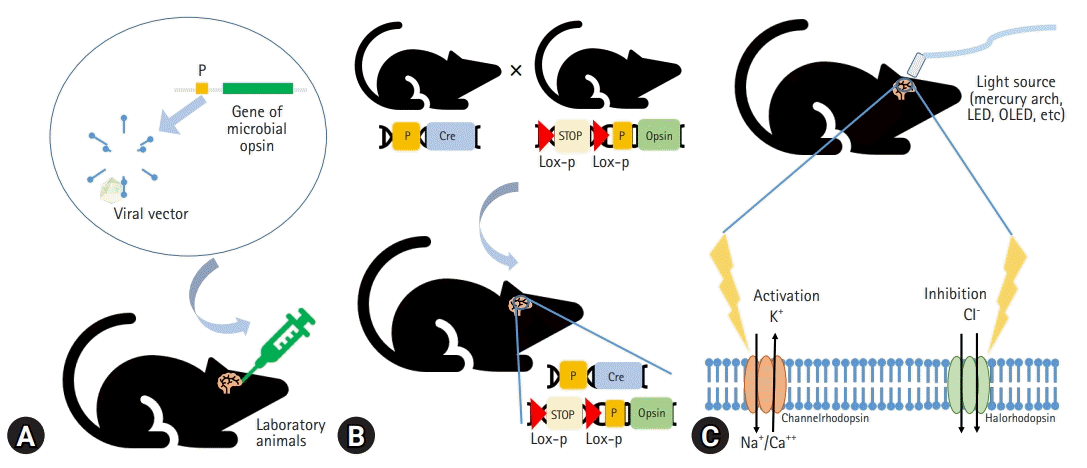
Based on this historical background, the application of optogenetics is fundamentally composed of (1) light-sensitive microbial opsin engineering, (2) genetic methods to introduce the opsin into cells, and (3) optical instruments for guiding light to activate or inhibit specific neural circuits to manipulate their behavior with temporal precision [23,25,31] (Fig. 1).
Light-activated proteins are required for the optical manipulation of molecular or cellular activity. Channelrhodopsin and anion-conducting channelrhodopsins are used to excite and inhibit neurons, respectively [32]. Halorhodopsin, bacteriorhodopsin, and archaerhodopsins are also used to inhibit neuronal activity [33,34].
The expression of microbial opsins in mammalian cells has been challenging. The use of viral vectors such as adeno-associated virus (AAV) is a fundamental method to express high levels of opsins, and the transfected neurons become electrically active in response to light [35,36]. Transgenic mice, including those using the Thy1 promoter, express opsins in the affected region at higher specificity than viral vectors do [36,37]. Using the Cre/lox recombinase system to create transgenic mice is a novel approach to optogenetics [36]. Photo-activable Cre recombinase can stably modify gene expression in the mouse brain [38,39].
Optogenetics principally depends on light stimulation. Although mercury arc lamps, light-emitting diodes (LEDs), and lasers have been used as in vitro light sources, organic LEDs are emerging technologies for optogenetics. Organic LEDs are suitable for implantation into the brain because they are softer, thinner, and more flexible than existing light sources and can supply adequate optical power over an acceptable temperature range [40]. Eventually, optogenetic techniques allow localized modulation of cell types of interest and simultaneous bidirectional control [41]. Moreover, the amplitude of stimulation and the time course are easily controlled by the light. This stimulation was shown to be relatively reproducible [42].
Gamma oscillation entrainment and Alzheimer disease
The different cell types in the central nervous system interact with each other, resulting in specific rhythmic oscillations such as delta, theta, alpha, beta, gamma, and sharp-wave ripples [6]. Jasper and Andrews [43] first introduced the term “gamma wave” in their report on the “normal differentiation of occipital and precentral regions in man” to describe higher frequencies (35–48 Hz) beyond the beta range. The widely reported frequency of gamma oscillations is 25 to 140 Hz, with the 40-Hz frequency being of particular interest [44]. In addition to light, sound, or tactile stimuli [44], various methods to stimulate gamma waves, including temporal interference [45], ultrasound stimulations [46], and optogenetics [47] have been suggested. Gamma oscillations correlate with various functions of the brain, including sensory processing and cognitive functions such as learning and memory [48]. Inter-areal coherence and local regulation have generated interest in gamma oscillations [49,50]. Parvalbumin-positive inhibitory neurons are dominant in gamma oscillation generation [6,51], while the activation of pyramidal neurons increases lower frequency oscillations in vivo [49].
Decreased synchronization of gamma oscillations [52-54] or enhanced gamma band power and lagged gamma responses [55,56] have been observed in patients with AD. Disturbances of slow gamma oscillations have also been reported in rodent AD models [57]. Interestingly, the transgenic APP-presenilin-1 mouse model of AD exhibits decreased gamma oscillation power in the lateral entorhinal cortex, which transmits various sensory inputs to the hippocampus and thus participates in memory processes analogous to those affected by human AD [58]. Decreased hippocampal slow gamma oscillation power has also been observed in a transgenic mouse model of AD [57].
Stimulation of gamma oscillations may have therapeutic potential for AD. Stimulation with light and sound sources at 1 to 30 Hz increases physical and cortical performance in patients with AD [59]. Light and sound stimulation between 8 and 15 Hz in patients with AD who are elderly improves cognitive performance, memory function, and alpha waves [44,60]. Visual stimulation by light flashes increases gamma band activity, in which patients with AD demonstrate increased frontoparietal gamma coherence and reduced occipitoparietal coherence [44,56].
Although the precise molecular and cellular mechanisms by which gamma oscillation stimulation ameliorates AD pathology are unknown, a correlation between Aβ and altered gamma oscillations has been reported. Decreased gamma oscillations could appear without Aβ plaques in TAS10 mice overexpressing human APP [61]. A close association between reduced gamma activity and functional behavioral deficits, as well as altered hippocampal gamma oscillations connected to Aβ, was found in the olfactory network of APPswe transgenic mice [62].
Optogenetic neuromodulation and Alzheimer disease
As the control of neural activity and neural circuit interrogation was made possible using optogenetic techniques [35,63], optogenetic approaches to AD subsequently began.
Since the loss of α4β2 nicotinic receptors is increased in AD [64-67], acetylcholine is released synaptically by optogenetic stimulation [68]. Bell et al. [68] suggested that activation of α4β2 receptors mediates nicotinic excitatory postsynaptic potential (EPSP) in CA1 interneurons by affecting the stratum lacunosum-moleculare using retroviral AAV expressing oChIEF in a Cre-dependent manner. Optogenetic activation of pyramidal neurons in the entorhinal cortex layer III improves synaptic defects between pyramidal neurons and CA1 parvalbumin-positive neurons in transgenic AD mice. It also halts the decrease in spatial learning and memory [69]. Although AAV has been generally used as a viral vector, the incidence of sharp wave ripples is reduced by optogenetic stimulation at the target location. The medial septum cholinergic stimulation of sleeping animals decreases sharp-wave ripples and advances theta-gamma oscillations. This research highlights the significance of the timing of cholinergic input. This could explain the limited success of cholinesterase inhibitor drugs in AD [70].
Optogenetic inhibition of hilar GABAergic interneurons of the dentate gyrus (DG) through Cre-dependent gene expression of enhanced halorhodopsin disrupts spatial learning and memory retrieval without affecting short-term working memory, motor coordination, and memory retention. Using optogenetic stimulation, GABAergic interneurons can be activated without affecting pyramidal neurons in the CA3 and CA1 regions [71]. Optogenetic stimulation of hippocampal memory engram cells in transgenic AD mice overexpressing APP/presenilin-1 induces memory retrieval. Optogenetic stimulation of DG engram cells improved long-term memory and spine density [72]. Optogenetic stimulation of the DG in APP/presenilin-1 × ArcCreERT2 × channelrhodopsin-2-enhanced yellow fluorescent protein mice improved memory impairment. Stimulation of DG neural ensembles leads to enhancement of memory retrieval and reactivation of neural ensembles [73], which suggests that optogenetic DG manipulation could be a target for AD treatment.
Optogenetic activation of glutamatergic neurons in Aβ-injected mice improves working memory and short-term memory without affecting long-term memory in the bilateral DG. This stimulation downregulates Aβ and upregulates neuronal nuclei, which are biomarkers of neuroprotection [10]. As antagonism of adenosine A2A receptor (A2AR) mimics memory impairment prevention in AD animal models [74-77], optogenetic activation of a chimeric rhodopsin-adenosine A2AR protein activates cyclic adenosine monophosphate (cAMP) signaling, which increases cAMP levels and mitogen-activated protein kinase phosphorylation. This activation induces memory dysfunction in the hippocampus through phospho-CREB signaling [77]. These reports suggest that multiple, targeted optogenetic approaches can be used to treat AD [10].
Optogenetics-induced gamma oscillations and Alzheimer disease
Since the excitation of gamma oscillations reduces circuit noise and amplifies signals that result in an increase in the signal transmission of the neocortex [49], optogenetics-induced gamma oscillations may have therapeutic potential for AD. Studies on the applications of optogenetics to 40-Hz gamma oscillations have been ongoing since the optogenetic stimulation of fast-spiking parvalbumin-positive interneurons in gamma oscillations was first demonstrated in mice [78]. Entrainment or synchronization of hippocampal gamma oscillations and spiking to 40 Hz via noninvasive stimuli, such as flashing lights or pulses of sound [79], reduces the Aβ load and activates microglia in a well-established 5XFAD mouse model of AD [80].
Decreased amyloidogenesis and increased amyloid endocytosis can be mediated by microglia [80]. Co-localization of microglia and Aβ was confirmed by histological analysis and induction of genes related to morphologic transformation of microglia was confirmed by gene expression profiling. That study suggested a neuroprotective role of gamma oscillations that affect neurons and microglia. Gamma oscillations also decrease phosphorylated tau protein levels [80].
In the JA20 AD mouse model, optogenetic stimulation of parvalbumin-positive interneurons restores slow gamma oscillations and increases spatial memory [47]. Accumulation of Aβ1-42 oligomers disrupts long-term potential and theta-nested gamma oscillations in the hippocampus. Furthermore, stimulation of GABAergic interneurons reduces neuroinflammation and activates autophagy. Photostimulated APP/presenilin-1 mice showed a significant decrease in escape latency in the Morris water maze test, indicating that optogenetic stimulation ameliorates spatial learning [81]. Optogenetic modulation of channelrhodopsin-2-expressing parvalbumin-positive interneurons restores gamma oscillations and gamma oscillation-induced spike timing-dependent long-term potentiation [82]. This activation selectively increases spontaneous inhibitory postsynaptic currents at theta and gamma frequencies and restores Aβ-induced reductions [83].
However, activation of parvalbumin-positive neurons by 40-Hz optical stimulation in the basal forebrain increased Aβ1-42 levels. Accumulation of amyloid plaques was increased in the medial prefrontal cortex and the septal nuclei. These results indicate that the method of activation of gamma oscillations changes the modulation of Aβ plaques [84]. Optogenetic stimulation of double-frequency slow waves increased the disruption of calcium homeostasis by Aβ and induced synaptic spine loss [85]. Subsequent human clinical trials of gamma oscillation band stimulation have shown mild cognitive improvements in patients with AD who have been exposed to light, sound, or tactile stimuli in the 40-Hz range [44]. However, the precise molecular and cellular mechanisms by which gamma oscillation band stimulation ameliorates AD pathology are unknown.
Limitations and prospects of optogenetics
Various anti-Aβ therapies are ongoing in clinical trials, but effective drugs are still lacking [86]. Although optogenetic technology for AD could be a new therapeutic approach, the major limitation of optogenetics is the use of viral vectors to express microbial opsins in human cells. Using viral vectors for gene therapy is considered a risky method that has not been fully tested to date, since AAV may cause activation of innate immunity and systemic inflammatory responses in humans [87,88]. Current optogenetics is mostly invasive because of the implantation of optic fibers, and overheating that induces tissue damage may be caused by the light [89]. Optogenetic stimulation also increases neuronal DNA double-strand breaks in mice [90]. The inappropriate use of optogenetics may paradoxically induce AD. Five months of chronic optogenetic stimulation could increase the formation of Aβ [91] and the release of tau protein [92]. Moreover, it remains a challenge to target opsins to defined organelles, including the plasma membrane or mitochondria [93,94] or to specific regions including dendrites or axon terminals [94].
Although optogenetics may have limitations, optogenetic neuromodulation allows for deep brain stimulation. In addition to AD, optogenetics-driven research has led to insights into Parkinson disease [93,95], autism, schizophrenia, drug abuse, anxiety, and depression [34,49,78,96]. As shown in the historical timeline (Fig. 2), this technology could modulate specific targets and neuronal activity [97]. The technical development of light delivery sources is also required. MicroLED arrays selectively stimulate opsins and act as biological amplifiers [98]. For in vivo modulation, the wireless form of a light source improves the application of optogenetics. Wireless control of light sources has been studied since 2011 [99]. In vivo injectable instruments require safe injectable battery technologies. The battery-free wireless system developed by Zhang et al. [100] could be another solution.
Conclusion
A new clinical approach for AD is needed because of the failure of Aβ monoclonal antibodies. Optogenetics could play key roles in learning the mechanisms of cellular responses and thus has the potential to treat neuronal diseases. In addition, optogenetics-induced gamma oscillations might provide a new method to modulate local neuronal signals in AD. Further research is needed to determine how optogenetics might be associated with gamma oscillations, and we suggest that, based on studies to date, it is highly related to the continuity of excitation-inhibition signals, frequency of gamma oscillations, and cytokine production-related cell signaling. Although optogenetics and gamma oscillations are currently not fundamental therapeutic approaches for AD, their combination could be a new way to manage AD. The development of actuators and sensors must precede the clinical use of optogenetics, since the viral vectors and opsins that have been used in optogenetic research are currently limited. As deep learning technology advances, the artificial manufacturing of opsins or modulation of viral vectors could be a breakthrough in optogenetic technology.

 XML Download
XML Download