

 PDF
PDF Citation
Citation Print
Print



INTRODUCTION
Hyperglycemia is synergistically caused by defects in the peripheral action of insulin (insulin resistance) and the inadequacy of insulin secretion by pancreatic beta cells (beta cell failure) [1]. Dysfunctional insulin secretion due to beta cell impairment is essential for the onset and progression of diabetes [2]. Various physiologic stressors create harmful environments that influence beta cell function. These stressors include chronic exposure to high levels of glucose and lipids, and increased insulin demand due to insulin resistance in peripheral tissues, both of which are commonly observed in obesity-associated type 2 diabetes (T2D). In harmful environments, beta cells initially perform compensatory mechanisms to handle the cellular stress, but ultimately the reactions become pathologic and induce beta cell dysfunction and apoptosis.
Although lipids are essential building blocks of cellular membranes and are mediators of signaling, free fatty acids (FFAs) are a harmful environmental factor that can cause stress for beta cells in pathologic conditions. Many studies have observed high levels of plasma FFAs in animal models of diabetes and in patients with T2D [3–5], and negative parameters such as glucose intolerance and impaired insulin release are related to increased levels of saturated fatty acids (FAs) [6–8]. Lipotoxicity due to chronic exposure to high levels of saturated FAs seriously damages beta cell function with reduced insulin synthesis [9–11] and secretion [12,13], and eventually induces apoptosis [14,15]. Studies on beta cell failure have reported various cellular mechanisms responsible for the lipotoxicity-induced dysfunction and apoptosis, but the mechanisms have not been fully described and new models of beta cell impairment are still being investigated [16]. The present review focuses on several mechanisms affected by lipotoxicity and discusses chemical means of preventing beta cell impairment.
SREBP-1c ACTIVATION AND DISORDERS OF LIPID METABOLISM
T2D is associated with high levels of FFAs, as well as accumulation of triglycerides in peripheral tissues [17]. Although FFAs can be cytotoxic in pancreatic beta cells by inducing apoptotic pathways that initiate mitochondrial dysfunction and cellular stress [18,19], the deposition of ectopic lipid in beta cells has been implicated as a main cause of beta cell impairment [20]. Chronic lipid stimulation of beta cells, such as in the later stages of T2D, appears to cause lipid accumulation and, ultimately, apoptosis [19,21]. Although the mechanism of beta cell apoptosis by FFAs is not completely understood, it is believed that lipotoxicity occurs when excess FFAs enter beta cells by non-oxidative metabolic pathways and cause lipid accumulation in beta cells [22,23].
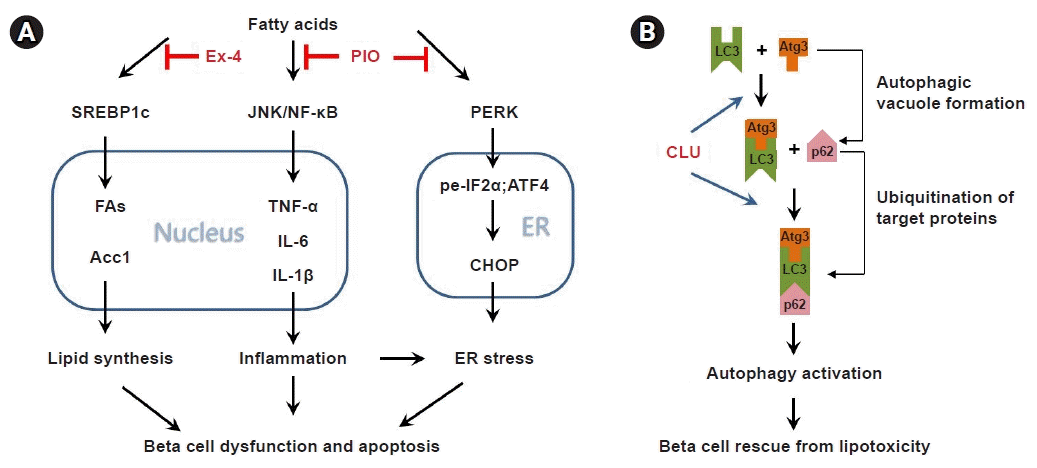
Sterol regulatory element-binding proteins (SREBPs) are essential transcription factors that regulate the expression of the genes involved in cholesterol and fatty acid synthesis [24]. Three isoforms of SREBPs have been identified (SREBP-1a, SREBP-1c, and SREBP-2). SREBP-1a and SREBP-1c are isoforms of SREBP-1 that are produced from a single gene via the use of alternative promoters and first exons. SREBPs are produced in the membrane of the endoplasmic reticulum (ER) following cleavage of the NH2-terminal domain, and the mature form moves to the nucleus and operates as a transcription factor [24,25]. SREBP-1 preferentially regulates the expression of genes associated with the biosynthesis of FFAs and triglycerides, whereas SREBP-2 mainly controls genes associated with cholesterol [26,27]. Significantly increased SREBP-1c expression has been observed in the islets and livers of animals in diabetes models [28]. Moreover, SREBP-1c activation partially blocked the effect of anti-diabetic molecules, including leptin, metformin, adiponectin, and thiazolidinedione (TZD) [29]. SREBP1c might be an important target in alleviating lipotoxicity. Our team demonstrated that SREBP1c is involved in the protective effect of exendin-4 (an incretin mimetic) against FFA-induced lipotoxicity in beta cells [30]. Treatment with an exendin-4 agonist dramatically decreased FFA-induced beta cell dysfunction and apoptosis by repressing SREBP1c expression and activity (Fig. 1A). Additionally, genetic ablation of SREBP1c significantly attenuated the FFA-induced lipotoxicity in insulin secretion and beta cell apoptosis. These results suggest that there is a strong correlation between lipotoxicity and SREBP1c, and that regulation of SREBP1c could be an effective means of preventing lipotoxicity in pancreatic beta cells.
INFLAMMATION AND ER STRESS
Several clinical studies have reported that anti-inflammatory therapy in patients with T2D improved beta cell function and hyperglycermia [31,32]. Exposure of pancreatic beta cells to circulating cytokines, such as tumor necrosis factor-alpha (TNF-α), interleukin 1-beta (IL-1β), and interferon-gamma, decreases insulin secretion and induces apoptosis by interruption of calcium (Ca2+) flow and cellular accumulation of islet amyloid polypeptide (IAPP) [32–35]. Inflammation of beta cells is also caused by lipotoxicity through activation of the c-Jun N-terminal kinase (JNK) pathway. Exposure to FFAs induces various cellular disorders such as ceramide formation, ER stress, and mitochondrial dysfunction, and the chronic cellular dysfunction associated with activated JNK [36]. Activated JNK signaling increases the expression of TNF-α, IL-1β, IL-6, and IL-8, and activates nuclear factor-kappa B (NF-κB), leading to beta cell dysfunction and apoptosis [35,37].
ER stress is associated with cellular damage in all tissues [38], and pancreatic beta cells are particularly susceptible due to characteristics of the biosynthesis and secretion of insulin. Irregular expansion of the ER was measured in beta cells from patients with T2D, and ER stress markers were increased in islets from db/db mice and patients with T2D [39,40]. These studies suggest that increased ER stress is an etiologic factor causing T2D via induction of beta cell failure. High levels of glucose and lipids in the blood are known to induce the overproduction of insulin, leading to beta cell dysfunction and apoptosis through ER stress.40 In beta cells, FFA-induced ER stress is promoted by the reduction of ER Ca2+ caused by mitochondrial dysfunction and a decreased influx of cellular Ca2+ [41]. In the early phase of ER stress, the unfolded protein response (UPR) occurs to maintain ER function by upregulation of UPR-associated proteins, such as glucose-regulated protein 78 (GRP78), phospho-eukaryotic translation initiation factor 2-α (p-eIF2α), and activating transcription factor 6 (ATF6). However, the chronic activation of UPR eventually induces excessive expression of C/EBP homologous protein (CHOP) and finally results in apoptosis. CHOP is a transcriptional regulator that stimulates cell death by downregulating the anti-apoptotic protein Bcl-2 and upregulating cellular reactive oxygen species and ER oxidoreductase 1, leading to hyper-oxidizing conditions in the ER [42].
In a recent study, the authors investigated the relationship between the inflammatory response and ER stress in pancreatic beta cells [43]. The expression of inflammatory cytokines and ER stress markers spontaneously increased in lipotoxicity-induced beta cells; therefore, we established a toxicity model to induce inflammation and ER stress. Treatment with lipopolysaccharide, an inducer of the inflammatory response, significantly increased the expression of TNF-α, IL-6, and IL-1β via JNK/NF-κB activation. The expression of ER stress markers also remarkably increased with lipopolysaccharide treatment. Although treatment with tunicamycin, an ER stress inducer, significantly increased the expression of ER stress markers, it did not impact the expression of inflammatory cytokines. These results suggest that an inflammatory response is an inducer of ER stress, but not vice versa, in pancreatic beta cells. We also investigated a protector from inflammation and ER stress. TZDs have mainly been reported to improve the insulin sensitivity of peripheral tissues via regulation of peroxisome proliferator-activator gamma (PPARγ) [44,45]. However, several recent studies in human and animal models also reported that TZDs protect pancreatic beta cells against lipotoxicity [46]. Treatment with TZDs improved the viability and function of human beta cells by protecting them from IAPP accumulation induced by lipotoxicity [47]. Using in vivo and in vitro models, we also showed that treatment with pioglitazone, a TZD, attenuated the lipotoxicity-induced inflammatory response and ER stress in beta cells [43]. Treatment with pioglitazone in mice fed a high-fat diet significantly reduced fasting blood glucose levels, as well as the expression of ER stress markers (ATF6 and GRP78) and monocyte chemoattractant protein (MCP1, a marker of the inflammatory response) in pancreatic islets. These results suggested that the protective effect of TZDs against lipotoxicity in pancreatic beta cells is mediated by repression of the inflammatory response and ER stress (Fig. 1A).
AUTOPHAGY
Autophagy is an essential process for the recycling of unnecessary cellular components and guaranteeing survival of the cell during starvation. Whether autophagy is harmful or beneficial to beta cell function and survival has been a matter of debate, but several recent studies suggest that autophagy is essential to maintain beta cell homeostasis and insulin secretion. Indeed, long-term treatment of beta cells with FFAs showed increased autophagosome numbers, and the activation of autophagosomes is an indicator of their protective role against FFA-induced cell death [48,49]. Exposure of pancreatic beta cells to interleukin-1 induced an accumulation of autophagic vacuoles, presenting severe damage in the late phase of autophagy [50]. Defective autophagy was also detected in the pancreatic islets of animals in a diabetes model (db/db mice, high-fat fed mice and Zucker rats) [48,51]. In addition, knockout of the beta cell specific autophagy-related 7 (Atg7) gene, which regulates autophagy, resulted in insulin deficiency and impaired glucose tolerance, suggesting that autophagy is essential to beta cell function and survival [52]. Consistently, our study showed that upregulation of autophagy via increased microtubule-associated protein light chain 3 (LC3) affinity protects beta cells from lipotoxicity-induced apoptosis [53]. We found that clusterin, a molecular chaperone, binds to LC3 and increases the affinity of LC3 to autophagy-associated factors, and treatment with clusterin significantly repressed beta cell apoptosis through facilitation of the autophagy process in palmitate-induced lipotoxicity conditions (Fig. 1B).
CONCLUSIONS
Pancreatic beta cell impairment is a critical risk factor for the onset and progression of T2D. Chronic exposure to FFAs causes lipotoxicity and is the leading cause of beta cell dysfunction, ultimately resulting in T2D. Therefore, a thorough understanding of the metabolism of lipotoxicity is essential for the prevention of beta cell failure and, ultimately, the treatment of T2D. This review discussed the intracellular metabolisms disturbed by lipotoxicity, including alteration of lipid metabolism, inflammatory response, ER stress, and autophagy. In addition, we discussed the possible role of several drugs in correcting broken cellular mechanisms and rescuing beta cells from lipotoxicity. This review supports an understanding of the molecular mechanisms of lipotoxicity-induced beta cell impairment and suggests attractive targets for therapies aimed at preventing beta cell failure, and ultimately treating T2D.
 XML Download
XML Download